Fibrin
Fibrin (lateinisch fibra ‚Faser‘, Faktor Ia der Blutgerinnungskaskade) ist der aktivierte, vernetzte „Klebstoff“ der plasmatischen Blutgerinnung. Es handelt sich um ein Protein, das durch die Einwirkung des Enzyms Thrombin (Faktor IIa der Gerinnungskaskade) aus der fadenförmigen löslichen Vorstufe, dem Fibrinogen (Faktor I der Gerinnungskaskade), gebildet wird. Das Fibrin polymerisiert anschließend und bildet ein Netz, das Blutgerinnsel („weißer Thrombus“), das die Wunde verschließt. Erst später wird durch Quervernetzung mithilfe des Faktors XIIIa der weiche Thrombus zu einer harten Kruste. Fibrin kann in Gegenwart von reinem Sauerstoff oder speziellen oxidativen Wundspüllösungen die Wundheilung beschleunigen.[1] Bei vielen chronischen Wunden entwickelt sich Fibrin allerdings aufgrund einer beständigen Entzündungsreaktion oft im Übermaß und bildet großflächige Beläge, die eine Abheilung behindern und mittels Débridement entfernt werden müssen.[2]
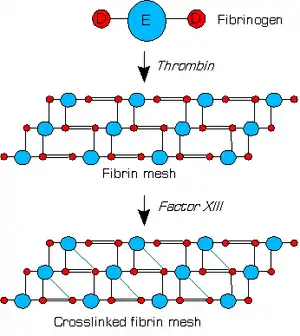
Details der Fibrinbildung
Bei der Bildung des weißen Thrombus wird zunächst durch Thrombin von der α-Kette des Fibrinogens das 16 Aminosäuren lange Fibrinopeptid A und von der β-Kette das 14 Aminosäuren lange Fibrinopeptid B abgespalten. Hierdurch wird die N-terminale Position an den Peptiden freigelegt und diese binden damit an der γ-Kette des Fibrinogens und lagern sich dort zu so genannten Protofibrillen zusammen. Im nächsten Schritt treten nun Protofibrillen zu mehr oder weniger dicken Fibrinfasern in einem Prozess zusammen, der als Lateralassoziation bezeichnet wird. Dabei kommt es auch zu Verzweigungen, so dass ein dreidimensionales Gebilde entsteht.[3][4]
Pathologie der Fibrinbildung
Die Gene FGA, FGB und FGG, welche die Unterketten des Fibrinogens codieren, können verschiedene Mutationen aufweisen, welche die Bildung von Fibrinogen und damit der Fibrinopeptide beeinträchtigen können. So sind Afibrinogenämien (kein Fibrinogen), Hypofibrinogenämien (verringerter Fibrinogengehalt des Blutes) und Dysfibrinogenämien (abnorme Fibrinogenmoleküle) bekannt.
Strukturveränderungen am Fibrinogen können verschiedene Defekte bei der Gerinnselbildung zur Folge haben:
- Störung der Freisetzung der Fibrinopeptide;
- veränderte Aggregation der Monomere;
- veränderte Lateralassoziation der Protofibrillen;
- Störung der späteren Vernetzung.
Etwa 60 Prozent der Defekte sind asymptomatisch. Bei den Symptomen dominieren Blutungsneigungen, in 15 Prozent gibt es eine Thromboseneigung. Erwähnenswert ist eine Gruppe von Defekten mit Einlagerung von Proteinaggregaten in Nieren und Milz (renale Amyloidose), wo die Nierenfunktionsstörung das Bild dominiert.[4]
Weblinks
Einzelnachweise
- A. Piaggesi, C. Goretti u. a.: A randomized controlled trial to examine the efficacy and safety of a new super-oxidized solution for the management of wide postsurgical lesions of the diabetic foot. In: The International Journal of Lower Extremity Wounds. Band 9, Nummer 1, März 2010, S. 10–15. doi:10.1177/1534734610361945. PMID 20207618.
- Kerstin Protz: Moderne Wundversorgung, 8. Auflage, Elsevier Verlag Urban & Fischer, München 2016, ISBN (Print) 978-3-437-27885-3, (e-Book) 978-3-437-17302-8, Seite 30.
- UniProt-Eintrag FGA.
- M. Meyer: Molekularbiologie der Gerinnung: Fibrinogen, Faktor VIII Hämostaseologie 24/-/2004 S. 108–115 Artikel als PDF.