Chronische myeloische Leukämie
Die chronische myeloische Leukämie (abgekürzt CML), auch chronisch-myeloische Leukämie und (seltener) chronische Myelose genannt, ist eine chronische Leukämie, die mit einer starken Vermehrung von Leukozyten (weißen Blutkörperchen), speziell von Granulozyten und ihren Vorstufen, im Blut und im blutbildenden Knochenmark einhergeht. Die Erkrankung ist in der Anfangsphase häufig symptomlos.
| Klassifikation nach ICD-10 | |
|---|---|
| C92.1 | Chronische myeloische Leukämie |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9863/3 | Chronische myeloische Leukämie o.n.A. |
| ICD-O-3 erste Revision online | |
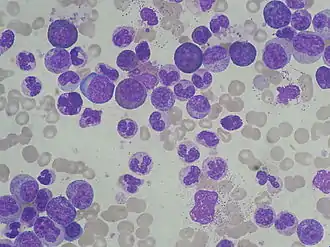
Die CML gehört zur Gruppe der myeloproliferativen Neoplasien (MPN), also Krankheiten, die aus einer (genetischen) Störung der im Knochenmark vorzufindenden hämatopoetischen Stammzellen resultieren. Bei der von Rudolf Virchow im Jahr 1845 beschriebenen und erstmals mit dem Namen Leukämie belegten Erkrankung handelte es sich mit hoher Wahrscheinlichkeit um eine CML.
Durch den Einsatz neuerer tumorspezifischer Medikamente, den sogenannten Tyrosinkinase-Inhibitoren, seit etwa der Jahrtausendwende haben sich die Prognose und die Behandlungsformen der CML deutlich gewandelt und die Erkrankung ist in vielen Fällen gut und verhältnismäßig nebenwirkungsarm behandelbar geworden. Die CML ist durch diesen Einsatz sogenannter zielgerichteter Therapien (englisch targeted therapies) geradezu zu einer Modellerkrankung für die ganze Hämatologie oder Tumortherapie im Allgemeinen geworden.[1][2]
Epidemiologie
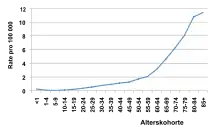
Die CML hat eine Inzidenz von etwa 1,6 Neuerkrankungen pro 100.000 Erwachsenen im Jahr und macht etwa 20 Prozent aller Leukämien aus. Sie ist ganz überwiegend eine Erkrankung des Erwachsenenalters und weist eine stetige Zunahme mit zunehmendem Alter auf. Männer sind etwa 1,4-mal häufiger betroffen als Frauen. Die Diagnose wird im Mittel (Median) mit 65 Jahren gestellt, etwa zehn Prozent der Patienten sind bei Diagnosestellung jünger als 35 Jahre.[3] Für ganz Deutschland schätzt man etwa 1.700 Neuerkrankungen pro Jahr, für die Schweiz kann man etwa 160 und für Österreich etwa 175 Neuerkrankungen pro Jahr annehmen.[5]
Ursachen und Entstehung
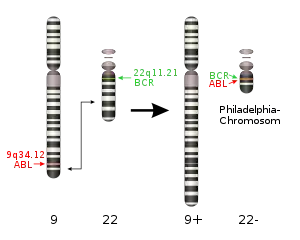
Ursache der Erkrankung ist die Veränderung und anschließende Proliferation (Vermehrung) einer einzigen multipotenten hämatopoetischen Vorläuferzelle. In fast allen Fällen findet man als Ursache eine reziproke Translokation zwischen den Chromosomen 9 und 22 (geschrieben: t(9;22)(q34;q11), oder abgekürzt t(9;22)). Bei beiden beteiligten Chromosomen ist der Bruchpunkt auf dem längeren Arm (q-Arm) lokalisiert, beim Chromosom 9 dichter zum Ende hin als beim Chromosom 22.
Der Chromosomenbruch liegt auf beiden Chromosomen im Bereich von Genen, nämlich ABL (oder ABL1) auf Chromosom 9 und BCR (breakpoint cluster region) auf Chromosom 22. Durch die Translokation kommt es zur Bildung von Fusionsgenen: BCR-ABL auf dem veränderten Chromosom 22 (mit dem Ende des Chromosoms 9) und ABL-BCR auf dem veränderten Chromosom 9 (mit dem Ende von Chromosom 22). Die Chromosomentranslokation ist zytogenetisch als verkürztes Chromosom 22, als sogenanntes "Philadelphia-Chromosom" sichtbar.
Das ABL-Gen kodiert für eine Tyrosinkinase und spielt eine wichtige Rolle bei der zellulären Wachstumsregulation. Kommt es zur Bildung des Fusionsgens BCR-ABL, dann ist ABL entscheidend in seiner Funktion gestört, die Tyrosinkinase-Aktivität ist dauerhaft aktiviert. Das BCR-ABL-Gen wirkt damit als Onkogen und führt zu einer gesteigerten und unkontrollierten Vermehrung der betroffenen Zelle. Die Translokation ist im Laufe des Lebens erworben und nicht vererbt oder vererbbar, da die Keimbahnzellen davon unberührt sind (soweit bekannt sind nur Zellen des blutbildenden Systems betroffen). Warum sie auftritt, ist nicht verstanden. Als Risikofaktoren für das Auftreten der Veränderung werden, wie bei Mutationen allgemein, ionisierende Strahlung oder Chemikalien diskutiert.[6]
Nicht bei allen CML-Patienten ist ein Philadelphia-Chromosom nachweisbar. In einer großen prospektiven Studie des Medical Research Councils konnte bei etwa 15 Prozent der Fälle zytogenetisch kein Philadelphia-Chromosom nachgewiesen werden.[7] Bei zwei Dritteln dieser Philadelphia-Chromosom-negativen Patienten war jedoch das BCR-ABL-Onkogen nachweisbar und das klinische Bild unterschied sich nicht wesentlich von dem der Philadelphia-Chromosom-positiven Patienten. Diese Patienten hatten zum Teil komplexe Chromosomenveränderungen, die die Translokation t(9;22) maskiert hatten. Bei einem Drittel, das heißt bei etwa fünf Prozent aller CML-Patienten, ergab sich kein Nachweis der t(9;22)-Translokation oder der BCR-ABL-Fusion. Das klinische Bild dieser Form unterschied sich deutlicher von der „typischen“ CML und hatte eine deutlich schlechtere Prognose.[7]
Verlauf
Die CML verläuft klassischerweise in drei Krankheitsphasen, der chronischen Phase, der Akzelerationsphase und der Blastenkrise (oder Blastenschub). Vor dem Aufkommen neuerer Medikamente und bevor die Option einer Stammzelltransplantation von Zellen gesunder Spender gegeben war, nahm der Krankheitsverlauf geradezu regelhaft diesen Verlauf. Die Blastenkrise war dabei das Endstadium der Erkrankung und führte zum Tod des Erkrankten. Auch heute ist nicht sicher vorauszusehen, wann bei einem Patienten die akzelerierte Phase und wann die Blastenkrise auftritt. Es sind Einzelfälle von CML-Patienten bekannt, die mehr als 20 Jahre in chronischer Phase gelebt haben. Bei anderen Patienten trat die akzelerierte Phase und Blastenkrise schon kurz nach Diagnosestellung auf. Heute sind Blastenkrisen seltener geworden, da gute Behandlungsmöglichkeiten für die chronische Phase bestehen. Gelegentlich werden sie aber immer noch diagnostiziert (häufig als „Erstmanifestation“ der CML).
Chronische Phase
Der Krankheitsbeginn ist langsam und schleichend und fällt oft über Jahre nicht auf. Leitsymptome dieser Phase sind die Leukozytose (Vermehrung der weißen Blutzellen) und die Splenomegalie (Milzvergrößerung). Die Splenomegalie erklärt sich durch die zunehmende Verdrängung der gesunden Blutbildung aus dem Knochenmark, so dass es zur extramedullären Blutbildung in Milz und später auch Leber kommt. Meist wird die Diagnose in diesem Stadium gestellt und nicht selten handelt es sich um eine Zufallsdiagnose, zum Beispiel anhand einer Blutbilduntersuchung, bei der eine Leukozytose auffällt. Im Differentialblutbild finden sich neben reifen Granulozyten unreife Vorstufen der myeloischen Reihe bis hin zu den Myeloblasten (sogenannte pathologische Linksverschiebung). Der Anteil an ganz unreifen Zellen (Blasten) liegt aber unter zehn Prozent.
Akzelerationsphase
Die Akzelerationsphase (lat. accelerare = beschleunigen) ist eine Übergangsphase zwischen der chronischen Phase und dem Blastenschub, in der die Erkrankung an Dynamik gewinnt. Sie ist durch zunehmende Leukozytose, Anämie (Blutarmut), Thrombozytopenie (Mangel an Blutplättchen) und eine zunehmende Milzschwellung gekennzeichnet. Im Differenzialblutbild zeigt sich ein Anteil von Blasten von zehn bis 30 Prozent. Die zytogenetische Analyse zeigt häufig zusätzlich zum Philadelphia-Chromosom neu aufgetretene Chromosomenveränderungen (am häufigsten: Isochromosom 17, ein zweites Philadelphia-Chromosom, Trisomie der Chromosomen 8 oder 19). Das subjektive Allgemeinbefinden des Patienten verschlechtert sich. Falls sich der Patient in chemotherapeutischer Behandlung befindet, zeigt sich eine abnehmende Medikamentenwirkung und Dosissteigerungen werden notwendig.
Blastenkrise
Die Blastenkrise tritt relativ plötzlich nach der Akzelerationsphase oder auch direkt aus der chronischen Phase heraus auf. In der Blastenkrise ändert die Erkrankung ihren Charakter von einem chronischen, eher langsamen Verlauf zu einem Verlauf, der dem einer akuten Leukämie entspricht. Die Blastenkrise ist definiert durch:
- Anteil der Blasten im peripheren Blut und/oder im Knochenmark ≥ 30 Prozent (nach Kriterien der Deutschen CML-Studiengruppe[6]) bzw. ≥ 20 Prozent (nach WHO-Definition), oder/und
- zytologisch oder histologisch gesicherte blastäre Infiltrate (Ansammlung von größeren Mengen an CML-Zellen in Geweben) außerhalb von Knochenmark, Milz oder Lymphknoten. Derartige Infiltrate bezeichnet man auch als Chlorome.[8]
Die Blasten bei etwa zwei Drittel aller Blastenkrisen zeigen einen myeloischen oder ganz undifferenzierten Immunphänotyp. Das restliche Drittel weist einen lymphatischen Immunphänotyp auf.[6] Im letzteren Fall kann die Blastenkrise ohne Kenntnis der Vorgeschichte meist nur schwer oder gar nicht von einer Philadelphia-Chromosom-positiven akuten lymphatischen Leukämie unterschieden werden. Einen gewissen Hinweis geben die verschiedenen mRNA-BCR-ABL-Transkripte, deren relative Verteilung bei CML und ALL unterschiedlich ist. Das Stadium der Blastenkrise endet unbehandelt relativ rasch (innerhalb von Wochen) tödlich.
Diagnosestellung
a) Knochenmark-Zytologie: Typisch für die CML ist eine Steigerung aller vier Zellreihen der Myelopoese: Thrombopoese (Thrombozyten-Bildung), Granulopoese (Granulozyten-Bildung), Monopoese (Monozyten-Bildung) und Erythropoese (Erythrozyten-Bildung). Es zeigt sich also in der Regel ein „volles (das heißt zellreiches Knochen-)Mark“. Die Granulozytopoese ist dabei am stärksten gesteigert und gewinnt in der Spätphase der Erkrankung auf Kosten der anderen Zellreihen die Oberhand. Dabei finden sich aber quantitative und qualitative Veränderungen mit manchmal ungewöhnlich kleinen Megakaryozyten ("Mikromegakaryozyten"), häufig auch einer Vermehrung von Basophilen und/oder Eosinophilen.
b) Zytogenetik und Molekulargenetik: In 95 Prozent Nachweis des Philadelphia-Chromosoms und/oder des BCR-ABL-Onkogens im Blut und Knochenmark. Ein Fehlen dieser Veränderungen ist prognostisch ungünstiger. Der Nachweis von BCR-ABL bei einer myeloproliferativen Erkrankung ist beweisend für die Diagnose einer CML. Manchmal wird das Philadelphia-Chromosom auch bei der akuten lymphatischen Leukämie gefunden.
c) Blutbild und klinisch-chemische Parameter des Blutes: Thrombozytose (in der Anfangsphase) bei einem Drittel der Patienten (Thromboserisiko!), Neutrophilie mit ausgeprägter Eosino- und Basophilie und Linksverschiebung (Vermehrung unreifer Vorstufen der Granulozyten) sowie mäßige Anämie. In der chronischen Phase finden sich aber trotz stark erhöhter Leukozytenzahlen nur wenige Blasten im Gegensatz zur akuten myeloischen Leukämie. Bei der CML können Leukozytenzahlen bis zu über 500.000/µl (Normalwert < 10.000/µl) erreicht werden. Zudem zeigen die CML-Zellen im Gegensatz zum Beispiel zu dem lymphozytären Zellen bei der chronischen lymphatischen Leukämie (CLL) oder Prolymphozytenleukämie (PLL), bei denen ebenfalls hohe Leukozytenzahlen im Blut erreicht werden, die Tendenz zur Adhäsion aneinander („Klebrigkeit“). Daher besteht bei solchen hohen Werte die akute Gefahr von Leukostase-Symptomen, das heißt die Fließfähigkeit des Blutes ist nicht mehr gewährleistet. Dadurch können in diesem Stadium Thrombosen und Durchblutungsstörungen auftreten (Venenthrombose der Netzhaut, Infarkt der Milz, schmerzhafter Priapismus durch Thrombose der Schwellkörper, Hirninfarkt, Myokardinfarkt).
d) Zytochemie: Der ALP-Index als Ausdruck der Aktivität der alkalischen Leukozytenphosphatase in den Neutrophilen ist meist erniedrigt. Diese Bestimmung hat allerdings heutzutage fast ganz ihre Bedeutung verloren.
Als Ausdruck des gesteigerten Zellumsatzes sind die Aktivität der Lactatdehydrogenase (LDH) sowie der Harnsäurespiegel im Blut meist erhöht.
Therapie
Allgemeine Therapieziele
Ziel der Therapie ist ein möglichst weitgehendes Zurückdrängen der Erkrankung bei vertretbaren Nebenwirkungen. Um die Effektivität der Behandlung zu messen, werden die folgenden Begriffe verwendet:
- hämatologisches Ansprechen (hematological response), das heißt Grad der Normalisierung des Blutbildes und Rückbildung der Splenomegalie; das hämatologische Ansprechen wird durch die klinische Untersuchung und das Differentialblutbild bestimmt;
- zytogenetisches Ansprechen (cytogenetic response), d. h. Prozentsatz der Zellen, in denen zytogenetisch das Philadelphia-Chromosom Ph+ nachweisbar ist;
- molekulares Ansprechen (molecular response), das heißt die Messung der BCR-ABL-mRNA mittels quantitativer Polymerase-Kettenreaktion (PCR)
Folgende Begriffe sind im internationalen medizinischen Sprachgebrauch definiert:[9]
Hämatologisches Ansprechen
Als Vollständiges hämatologisches Ansprechen (complete hematological response, CHR) wird die vollständige Normalisierung des Blutbilds und der klinischen Symptome bezeichnet, d. h. Thrombozyten unter 450 × 109/l, Leukozyten unter 10 × 109/l, ein Differentialblutbild ohne unreife granulozytäre Vorstufen (Myelozyten, Promyelozyten, Myeloblasten) und mit weniger als fünf Prozent Basophilen sowie keine tastbare Splenomegalie.
Zytogenetisches Ansprechen
Die Messung des zytogenetischen Ansprechens erfolgt mittels zytogenetischer Untersuchung. Dabei wird der Prozentsatz der Metaphasen mit nachweisbarer Philadelphia-Translokation ermittelt.
- Vollständiges zytogenetisches Ansprechen (complete cytogenetic response, CCyR): Ph+ 0 Prozent
- Partielles zytogenetisches Ansprechen (partial cytogenetic response, PCyR): Ph+ 1–35 Prozent
- Geringes zytogenetisches Ansprechen (minor cytogenetic response, MiCyR): Ph+ 36–65 Prozent
- Minimales zytogenetisches Ansprechen (minimal cytogenetic response): Ph+ 66–95 Prozent
- Kein zytogenetisches Ansprechen (no cytogenetic response): Ph+ >95 %
Molekulares Ansprechen
Die Messung des molekularen Ansprechens erfolgt mittels RT-PCR. Um die Vergleichbarkeit zwischen verschiedenen Laboren zu gewährleisten, wurde eine internationale Skala (IS) entwickelt.
- Vollständiges molekulares Ansprechen (complete molecular response, CMR): BCR-ABL nicht detektierbar
- Gutes molekulares Ansprechen (major molecular response, MMR): BCR-ABL ≤ 0,10IS
Vereinfacht kann man sagen, dass bei einem vollständigen hämatologischen Ansprechen der Prozentsatz der CML-Zellen im Blut/Knochenmark höchstens im unteren zweistelligen Prozentbereich liegen kann, bei einem vollständigen zytogenetischen Ansprechen liegt er höchstens im unteren einstelligen Prozentbereich und bei einem vollständigen molekularen Ansprechen liegt er unterhalb des Promillebereichs.
Hydroxycarbamid
Die Normalisierung der Leukozytenzahl lässt sich oft bereits durch Anwendung von Hydroxycarbamid (Hydroxyharnstoff, englisch: Hydroxyurea, HU) erreichen. Dieses Zytostatikum ist seit langem auf dem Markt. Es hemmt die Umwandlung der Ribonukleotide in Desoxyribonukleotide und ist auch bei den anderen oben genannten myeloproliferativen Erkrankungen wirksam. Typische Dosierungen sind eine Tablette zu 500 mg bis etwa vier Tabletten (= zwei Gramm) täglich.
Nebenwirkungen sind selten, gelegentlich leichte Übelkeit, sehr selten Schleimhautschäden oder Lebertoxizität. Durch diese Therapie ist es möglich, die Leukozytenzahl im Blut annähernd wieder auf Normalbereiche zu reduzieren (5 000 bis 10 000/μl). Ein nachhaltiger Einfluss auf den Krankheitsverlauf ist jedoch mit dieser Normalisierung nicht verbunden. Nach durchschnittlich drei Jahren kommt es zur weiteren Verschlechterung des Zustandes und zum Eintritt in die Akzelerationsphase. Das mediane Überleben von Patienten, die mit Hydroxycarbamid behandelt werden, ist etwas länger als das von ganz unbehandelten Patienten und betrug in historischen Therapiestudien ungefähr 4 ½ Jahre.[10]
Der Einsatz von Hydroxycarbamid bei der CML ist heute nur noch in bestimmten Situationen sinnvoll, beispielsweise dann, wenn in der Anfangsphase der Behandlung sehr hohe Leukozytenzahlen reduziert werden sollen, oder wenn der Einsatz von TKI (siehe unten) aufgrund untolerierbarer Nebenwirkungen nicht möglich ist, oder wenn der Patient nur noch eine sehr kurze Lebenserwartung aufgrund anderer bestehender Krankheiten (z. B. fortgeschrittene Krebserkrankung) hat.
Interferon-α
Interferon alpha (α) (IFN-α) ist ein Zytokin, welches von Leukozyten gebildet und freigesetzt wird; es dient der Immunantwort auf virale und bakterielle Infektionen und induziert in den Zielzellen, zum Beispiel bei Virusbefall, unter anderem eine Proliferationshemmung. Zudem steigert es die Aktivität der zytotoxischen T-Zellen und Makrophagen. Interferon wird in der Regel 3×/Woche subkutan injiziert. Typische Dosierungen sind 3×0,5 bis 3 × 6 Millionen Einheiten pro Woche. Es kann entweder als Monotherapie oder in Kombination mit anderen Medikamenten gegeben werden. Interferon-α ist ein häufig nicht sehr gut verträgliches Medikament. Es kann zu grippeähnlichen Symptomen kommen, die aber abhängig von der Dosis auftreten. Die Interferon-Therapie hat in vielen Fällen Auswirkungen auf die Konzentrations- und Merkfähigkeit, Depressionen können auftreten, sowie Schwindel, Verwirrtheit und Polyneuropathien. Zudem hat die Behandlung Auswirkungen auf den Verdauungstrakt und auf die Leber.
Tyrosinkinase-Inhibitoren (TKI)
Bei Patienten, die BCR-ABL-positiv sind, kann eine spezifische Therapie zum Einsatz kommen. Seit etwa der Jahrtausendwende gibt es Medikamente, die spezifisch die Tyrosinkinase-Enzymaktivität des ABL-Anteils von BCR-ABL kompetitiv hemmen können. Diese Tyrosinkinase-Inhibitoren haben die Behandlung der CML geradezu revolutioniert und zu einer dramatischen Verbesserung der Behandlungsergebnisse geführt. Man unterscheidet Tyrosinkinase-Inhibitoren (TKI) der ersten, zweiten und dritten Generation, die nacheinander auf den Markt gekommen sind. Der wichtigste TKI der ersten Generation ist Imatinib. Die Substanz erhielt im Jahr 2001 bzw. 2003 die Zulassung für die Behandlung der CML in Europa bzw. in den Vereinigten Staaten. TKI der zweiten Generation sind Nilotinib und Dasatinib, TKI der dritten Generation sind Bosutinib und Ponatinib. Als weiterer TKI ist seit 2021 der STAMP-Inhibitor Asciminib verfügbar, der nicht an der ATP-Bindungsstelle des BCR-ABL-Fusionsproteins angreift, sondern an der Myristoyl-Bindungstasche.
Tyrosinkinase-Inhibitoren gelten heute als medikamentöser „Goldstandard“ der CML-Behandlung.
Imatinib und die IRIS-Studie
Die Effektivität und Überlegenheit von Imatinib gegenüber allen bislang bekannten anderen medikamentösen Behandlungsformen wurde in einer großen internationalen Phase-III-Therapiestudie mit mehr als 1000 CML-Patienten, der sogenannten IRIS-Studie,[11] deutlich (IRIS. = International Randomized Study of Interferon and STI571). Ursprünglich war die IRIS-Therapiestudie als zweiarmige Vergleichsstudie geplant, d. h. die Hälfte der Patienten sollte Imatinib als Behandlung erhalten und die andere Hälfte der Patienten die bisherige Standardbehandlung aus Interferon-α und Cytarabin. Schon bei der ersten Zwischenauswertung[11] mit einer mittleren Beobachtungszeit von 18 Monaten wurde aber die Überlegenheit der Imatinib-Behandlung deutlich. Bei den mit Imatinib behandelten Patienten zeigte sich ein gutes zytogenetisches Ansprechen (siehe die Begriffsdefinitionen oben) von 87,1 Prozent und ein komplettes zytogenetisches Ansprechen von 76,2 Prozent. In der Interferon-α/Cytarabin-Gruppe waren die entsprechenden Zahlen 34,7 Prozent und 14,5 Prozent. In der Imatinib-behandelten Patientengruppe zeigten 96,7 Prozent kein Fortschreiten der Krankheit zur Blastenkrise/Akzelerationsphase, während es in der Interferon-α/Cytarabin-Gruppe 91,5 Prozent waren. Insgesamt wurde die Imatinib-Behandlung auch besser vertragen als die Behandlung mit Interferon α/Cytarabin.
Diese Ergebnisse führten dazu, dass die meisten Patienten aus der Interferon-α/Cytarabin-Gruppe in den Behandlungsarm mit Imatinib wechselten (dieser cross-over war im Rahmen der IRIS-Studie erlaubt). Außerdem erfolgte (wesentlich aufgrund der Ergebnisse der IRIS-Studie) die beschleunigte Zulassung von Imatinib zur Behandlung der CML durch die amerikanische Food and Drug Administration[12] und die Europäische Arzneimittel-Agentur[13] und in der Folge verließen viele Patienten, die im Rahmen der IRIS-Studie mit Interferon-α/Cytarabin behandelt wurden, die Studie und führten die Behandlung außerhalb der Studie mit Imatinib fort. Die IRIS-Studie hat sich daher mittlerweile von einer anfänglich zweiarmigen Studie praktisch in eine reine Beobachtungsstudie für die Behandlung von CML-Patienten mit Imatinib gewandelt.
In einer im Jahr 2009 veröffentlichten Aktualisierung der IRIS-Studiendaten zeigte sich dabei bei einer medianen Beobachtungszeit von acht Jahren bei den von Anfang an mit Imatinib behandelten Patienten ein progressionsfreies Überleben von 92 Prozent der Patienten.[14]
Imatinib wurde nach der Bekanntgabe der Ergebnisse der IRIS-Studienergebnisse zum medikamentösen Standard der CML-Behandlung. Über die optimale Imatinib-Dosis besteht allerdings noch kein Konsens. Die übliche Dosis sind 400 mg täglich. Die deutsche CML IV-Therapiestudie untersuchte, ob eine 800 mg-Tagesdosis toleriert wird und bessere Ergebnisse erbringt. Nach ersten Auswertungen scheint es so zu sein, dass ältere Patienten (≥ 65 J.) mit einer 800 mg Imatinib-Medikation deutlich schneller eine Remission erreichen, als mit einer 400 mg-Medikation, was allerdings auch mit höheren Nebenwirkungen erkauft werden muss. Die Schlussfolgerung aus dieser Analyse war die, dass die Imatinib-Dosis bei Patienten ≥ 65 J. möglichst über 400 mg täglich liegen sollte.[15] Gesichert erscheint, dass die Tagesdosis allgemein nicht wesentlich unter 400 mg liegen sollte, da dadurch die Entwicklung von Resistenzen begünstigt wird. Mittlerweile hat sich die Situation allerdings dadurch gewandelt, dass neuere Tyrosinkinase-Inhibitoren der zweiten oder dritten Generation verfügbar sind (siehe unten). In den aktuellen Leitlinien wird kein spezieller TKI explizit empfohlen, es werden nur Vorgaben für die Therapieziele gemacht, die zu erreichen sind.[16]
Imatinib ist meist relativ gut verträglich. An Nebenwirkungen können Übelkeit und Erbrechen (selten), Ödeme, Pleura- und Perikardergüsse (Flüssigkeitsansammlung im Brustfell bzw. Herzbeutel), Transaminasenanstieg, Muskelkrämpfe und Hautausschläge auftreten.
Nilotinib und Dasatinib: TKI der zweiten Generation
Trotz der bemerkenswerten Behandlungserfolge mit Imatinib benötigen etwa 20 bis 25 Prozent der CML-Patienten im Laufe der Imatinib-Behandlung innerhalb von 5 bis 8 Jahren eine alternative Therapie.[17] Gründe hierfür sind eine primär nicht genügende Wirksamkeit, ein Wirkungsverlust von Imatinib, oder eine Imatinib-Unverträglichkeit. In dieser Situation gibt es zwei wesentliche Alternativen, zum einen der Wechsel zu einem anderen Tyrosinkinase-Inhibitor und zum anderen die Durchführung einer allogenen Stammzell- oder Knochenmarktransplantation. Ursache für den Wirkungsverlust kann eine Mutation im Bereich der Tyrosinkinase-Domänen des BCR-ABL-Gens sein.
Zwei neue Tyrosinkinase-Hemmstoffe der zweiten Generation, Dasatinib (Handelsname Sprycel) und Nilotinib (Handelsname Tasigna), wurden zur Behandlung der CML bei Imatinib-Resistenz oder -Unverträglichkeit zugelassen. Sowohl Dasatinib[18] als auch Nilotinib[19] sind in vitro deutlich wirksamer in der Hemmung der ABL-Kinase und wirken auch bei den meisten Imatinib-Resistenzen. Lediglich bei der Mutation T315I sind beide Substanzen wirkungslos. Dabei liegt eine Genmutation vor, die zum Aminosäureaustausch Threonin → Isoleucin an Position 315 führt. Isoleucyl-Reste sind erheblich größer als Threonyl-Reste und verhindern so möglicherweise die Bindung des Pharmakons an die spezifische Bindungsstelle.
Die im Jahr 2013 begonnene deutsche TIGER-Therapiestudie (TIGER = Tasigna/Interferon in Germany) setzt ganz auf Nilotinib (zum Teil in einer späteren Phase kombiniert mit Interferon α). Imatinib soll im Rahmen dieser Studie nur bei Nilotinib-Unverträglichkeit gegeben werden, während Dasatinib (oder die allogene Stammzelltransplantation) beim Auftreten einer Nilotinib-Resistenz zum Einsatz kommen sollen.[20]
TKI der dritten Generation
Weitere neuere TKI sind Bosutinib (Bosulif) und Ponatinib (Iclusig), die beide im Jahr 2012 in den Vereinigten Staaten zur Behandlung der CML bei Unverträglichkeit oder Unwirksamkeit anderer TKI zugelassen wurden. Einige Monate später erfolgte auch die Zulassung in Europa. Insbesondere Ponatinib ist von großem Interesse, da es auch gegen die BCR-ABL T315I-Mutation wirksam ist.[21]
Resistenzen gegen Tyrosinkinase-Inhibitoren
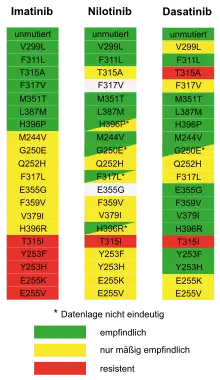
Bei der Therapie mit Tyrosinkinase-Inhibitoren tritt mitunter das Phänomen auf, dass sich die Wirkung des Medikaments zunehmend abschwächt und schließlich ganz verloren geht. Der früheste Indikator hierfür ist ein Anstieg des BCR-ABL-Niveaus (das üblicherweise unter Therapie alle drei Monate im Blut bestimmt wird). Falls nichts weiter unternommen wird, breitet sich die Erkrankung erneut aus und nimmt den oben geschilderten Verlauf. Man spricht in solchen Fällen von einer Resistenz gegen das Medikament, also zum Beispiel einer Imatinib-Resistenz oder einer Nilotinib-Resistenz.
Ursache für solche Resistenzen sind häufig Nukleotid-Mutationen im BCR-ABL-Onkogen, die zu Aminosäure-Austauschen führen. Das Medikament zeigt dann keine Wirkung mehr gegen das mutierte BCR-ABL-Protein. Man nimmt heute an, dass die meisten dieser Mutationen in geringer Menge schon bei Therapiebeginn vorhanden sind und dann durch die Therapie herausselektiert werden (nur der mutierte, d. h. resistente Klon kann wachsen). Manche der Mutationen treten wahrscheinlich aber auch zufällig während des Krankheitsverlaufs auf. Viele BCR-ABL-Mutationen wurden in den vergangenen Jahren charakterisiert. Von klinischem Interesse ist dabei der Umstand, dass manche BCR-ABL-Mutationen zwar gegen einen Tyrosinkinase-Inhibitor Resistenz verleihen, aber gegen andere empfindlich sind. In solchen Fällen ist dann ein Wechsel auf einen anderen Tyrosinkinase-Inhibitor sinnvoll. In einem Consensus-Statement einer Expertengruppe des European LeukemiaNet aus dem Jahr 2011 wurde beim Nachweis von Mutationen unter Imatinib-Therapie folgendes Vorgehen empfohlen:[26]
- T315I: allogene Transplantation oder experimentelle Therapie;
- V299L, T315A, V517L/V/I/C: eher Nilotinib als Dasatinib;
- Y253H, E255K/V, F359V/C/I: eher Dasatinib als Nilotinib;
- jede andere Mutation: Imatinib höherdosiert oder Dasatinib oder Nilotinib.
Es gibt allerdings auch das Phänomen der Resistenz, ohne dass eine BCR-ABL-Mutation nachgewiesen werden kann. Hier liegen dann offensichtlich andere genetische Veränderungen zugrunde, die bisher nicht gut verstanden sind. In dieser Situation kann auch ein Wechsel des Tyrosinkinase-Inhibitors sinnvoll sein oder eine Dosiserhöhung des bisher gegebenen.
Therapieziele bei der TKI-Therapie
Die Expertengremien wurden im Jahr 2013 Empfehlungen für die zu erreichenden Therapieziele formuliert.[16] Dabei wurde ausdrücklich kein bestimmter TKI (z. B. Imatinib, Nilotinib, Dasatinib, o. a.) empfohlen. Die Therapieziele bei der Behandlung mit Tyrosinkinase-Inhibitoren sind ein vollständiges hämatologisches Ansprechen (CHR) nach spätestens drei Monaten, ein vollständiges zytogenetisches Ansprechen (CCyR) nach spätestens sechs Monaten und eine weitgehende molekulare Remission (MMR) nach spätestens 18 Monaten. Falls sich Warnhinweise ergeben, wird empfohlen entsprechend häufigere Kontrollen durchzuführen (z. B. bis zu 1× monatlich BCR-ABL-Messung), falls ein Therapieversagen vorliegt muss die Therapie geändert werden.
| Kriterien zur Beurteilung der Wirksamkeit einer TKI-Therapie[16] | |||
|---|---|---|---|
| Zeit | Optimal | Warnhinweise | Therapieversagen |
| Diagnosezeitpunkt | Entfällt | Hochrisiko (s. u. Risikoscores), zusätzliche chromosomale Aberrationen in Ph+ Zellen |
Entfällt |
| 3 Monate nach Diagnose | BCR-ABL ≤ 10 % und/oder Ph+ Metaphasen ≤ 35 % |
BCR-ABL >10 % und/oder Ph+ Metaphasen 35 bis 95 % |
keine vollständige hämatologische Remission (CHR) und/oder Ph+ Metaphasen > 95 % |
| 6 Monate nach Diagnose | BCR-ABL ≤ 1 % und/oder Ph+ Metaphasen 0 % |
BCR-ABL 1 bis 10 % und/oder Ph+ Metaphasen 1 bis 35 % |
BCR-ABL >10 % und/oder Ph+ Metaphasen > 35 % |
| 12 Monate nach Diagnose | BCR-ABL ≤ 0,1 % | BCR-ABL 0,1 % bis 1 % | BCR-ABL > 1 % und/oder Ph+ Metaphasen > 0 % |
| zu jedem späteren Zeitpunkt | BCR-ABL ≤ 0,1 % (MMR) | zytogenetische klonale Aberrationen | Verlust einer kompletten zytogenetischen Remission und/oder Verlust einer hämatologischen Remission und/oder bestätigter (!) Verlust einer weitgehenden molekularen Remission (MMR) und/oder Nachweis von BCR-ABL-Mutationen |
Knochenmark- oder Blutstammzelltransplantation
Die allogene Stammzelltransplantation (SZT) (seltener auch Knochenmarktransplantation KMT) ist nach bisherigem Wissen die einzige Therapieform, die zu einer vollständigen Heilung führen kann. Es gibt zwar auch vereinzelt Patienten, bei denen die Therapie mit Tyrosinkinase-Inhibitoren zu einer Nicht-mehr-Nachweisbarkeit des BCR-ABL-Fusionsgens führt. Hier ist aber nicht klar, ob es sich um wirkliche Heilungen oder nur um ein Herunterdrücken der Erkrankung unter die Nachweisgrenze handelt. Da die Prognose am günstigsten ist, je früher die SZT/KMT durchgeführt wird, sollte so früh wie möglich darüber entschieden werden. Das Problem dieser Transplantation ist die relative hohe Mortalität durch Komplikationen (insbesondere schwere Infektionen) während der Transplantationsprozedur und das mögliche Auftreten von schweren Autoaggressionskrankheiten nach Transplantation (Graft-versus-host-Erkrankung, GvHD).
Dieser Effekt kann entstehen, weil sich aus den Stammzellen des Knochenmarkes alle Blut- und Abwehrzellen des Körpers bilden. Es wird also nicht allein die Blutbildung ersetzt, sondern wesentliche Teile des Immunsystems transplantiert. Während bei herkömmlichen Transplantationen ein host-versus-graft-Effekt (‚Empfänger gegen Transplantat‘), also eine typische Abstoßungsreaktion zu beobachten ist, richtet sich hier das transplantierte Immunsystem gegen den neuen Körper. Aus diesem Grund muss der SZT/KMT auch eine maximal aggressive Chemotherapie vorangehen, um das körpereigene Immunsystem möglichst vollständig zu zerstören. Die Etablierung der neuen Stammzellen könnte durch ein verbleibendes Immunsystem empfindlich gestört werden.
Der Graft-versus-Host-Effekt ist aber nicht nur nachteilig für den Patienten: ein Teil der Transplantierten profitiert möglicherweise von einer Bekämpfung der verbliebenen Tumorzellen durch das neue Immunsystem. Man nennt diesen Effekt graft-versus-leukemia-Reaktion (GvL-Reaktion).
Eine Transplantation wird nur durchgeführt bei:
- Patienten, bei denen eine Behandlung mit Imatinib oder neueren Tyrosinkinase-Inhibitoren unwirksam ist,
- jüngeren Patienten und
- Patienten, für die ein kompatibler Spender gefunden wird (Familien- oder Fremdspender).
Therapiekosten
| Medikament | Typische Dosierung pro Tag |
Tages- therapiekosten |
|---|---|---|
| Glivec | 400 mg | 116 € |
| Tasigna | 600 mg | 131 € |
| Sprycel | 100 mg | 169 € |
| Roferon A | 3× 6 Mio. IE (pro Woche) | 31 € |
| Litalir | 1 g | 4,52 € |
| Hinweis: Diese Tabelle soll nicht suggerieren, dass die aufgeführten Medikamente wirkungsäquivalent sind. Hier sind nur die Therapiekosten verglichen. Hinsichtlich der Wirksamkeit bestehen große Unterschiede. | ||
Das Standard-Medikament Imatinib steht in Tablettenform in Dosierungen von 100 mg oder 400 mg zur Verfügung. Die Standard-Dosierung sind 400 mg Imatinib täglich. Der deutsche Apothekenpreis (Herstellerempfehlung) für die Packung Glivec mit 90 Tabletten à 400 mg betrug 10.434,48 € (Stand: 01/2022, entspricht 116 € pro Tablette).[27] Sprycel-Packungen zu 30 Tabletten à 100 mg kosteten 5063,05 €, was bei der üblichen 100 mg Tagesdosis einem Tagestherapiepreis von 169 € entspricht.[27] Tasigna gab es in der Stärke 150 mg (112 Kapseln) zu einem Preis von ca. 3938,56 €,[27] was bei der üblichen Tagesdosis von 600 mg Tagestherapiekosten von 131 € mit sich zieht. Die Medikamentenkosten lagen damit für einen CML-Patienten zwischen etwa 41.369 € (Glivec), 61,727 € (Sprycel) und 48.848 € (Tasigna) pro Jahr.
Zum Vergleich: die Behandlung mit Hydroxycarbamid (Hydroxyurea, zum Beispiel Litalir) verursacht bei 1 g Tagesdosis jährliche Therapiekosten von ca. 1650 €, die mit Interferon alpha (zum Beispiel Roferon 3 × 6 Mio. IE wöchentlich) jährliche Kosten von ca. 11.500 €.[27]
Auf Kritik stieß der Umstand, dass mittlerweile fünf verschiedene gut wirksame Tyrosinkinase-Inhibitoren von verschiedenen Herstellern auf dem Markt sind (Stand Juli 2013: Imatinib und Nilotinib von Novartis, Dasatinib von Bristol-Myers Squibb, Bosutinib von Pfizer, Ponatinib von Ariad Pharmaceuticals), aber die Therapiekosten weiterhin exorbitant hoch sind und sogar noch weiter steigen.[28] In einem im Mai 2013 in der angesehenen amerikanischen Fachzeitschrift Blood erschienenen Artikel kritisierte ein internationales Kollektiv von mehr als 100 Experten deutlich die hohen Preise der modernen Tumormedizin, speziell am Beispiel der CML und wies darauf hin, dass derartige Preise auf die Dauer für kein Gesundheitssystem tragbar seien.[29] Auch in hochentwickelten Ländern wie den Vereinigten Staaten oder Schweden gäbe es eine signifikante Zahl an Patienten, die sich aufgrund der hohen Medikamentenkosten diese Therapie nicht leisten könnten und deswegen ein schlechteres Überleben hätten. Von den weltweit etwa 1,2 bis 1,5 Millionen CML-Erkrankten würden nur etwa 235.000 bis 250.000, also 20 bis 25 % mit dem Standardmedikament Imatinib behandelt, was wesentlich an den hohen Therapiekosten liege.[28] Außerdem wurde darauf hingewiesen, dass erhebliche Preisunterschiede zwischen verschiedenen Ländern bestünden. Als Jahrestherapiekosten von Imatinib wurden für verschiedene Länder angegeben (in 1.000 $US): USA 92, Deutschland 54, Großbritannien 34, Frankreich 40, Italien 31, Südkorea 29, Japan 43, Russland 24. Als Hauptgrund für die außerordentlich unterschiedlichen Kosten wurden die unterschiedlichen Vorgaben des Gesetzgebers ausgemacht, die zu unterschiedlichen Freiheiten in der Preisgestaltung des Herstellers führten.[29]
Auf erhebliches internationales Interesse stieß auch der Rechtsstreit zwischen der Firma Novartis und Indien über die Gültigkeit des Patentschutzes für Glivec (Imatinib) in Indien. Letztlich unterlag Novartis letztinstanzlich am 1. April 2013 vor dem obersten Gerichtshof in Indien, was zur Folge hatte, dass Imatinib in Indien als Generikum auch von anderen Herstellern erhältlich ist.[30][31]
Die Kosten für eine allogene Stammzelltransplantation werden inklusive der Nachbehandlung auf etwa 150.000 € pauschal geschätzt, wobei sich diese Zahlenangabe nur auf die unmittelbare Nachbehandlung bezieht. Treten später Komplikationen auf, wie eine chronische graft-versus-host-Erkrankung, können die tatsächlichen Gesamtkosten sehr viel höher liegen. Nicht selten verlieren transplantierte Patienten auch ihre Arbeits- und/oder Berufsfähigkeit, so dass die „volkswirtschaftlichen“ Gesamtkosten deutlich höher liegen.
Prognose
| Score | berücksichtigte klinische und Blut-Parameter |
Weblink zur Berechnung |
|---|---|---|
| EUTOS-Score | Milzgröße Basophile | eutos.org |
| Sokal-Hasford-Score | Alter Milzgröße Thrombozyten Basophile Eosinophile Myeloblasten | leukemia-net.org |
Überlebensraten im historischen Überblick
Bevor Medikamente zur Verfügung standen, war die CML die Erkrankung aus dem Formenkreis der Myeloproliferativen Neoplasien (MPN) mit der ungünstigsten Prognose. Das mediane Überleben von CML-Patienten betrug ohne Behandlung nur etwa drei bis vier Jahre. Hydroxycarbamid führte zu einer geringen Verbesserung auf ca. 4 ½ Jahre[10] und Interferon führte darauf folgend zu einer weiteren Verbesserung des medianen Überlebens auf etwa 5 ½ Jahre.[10]
Heute liegen die Fünf-Jahres-Überlebensraten bei Imatinib-Behandlung bei etwa 90 Prozent oder sogar noch darüber. Selbst bei mittlerweile mehr als 10 Jahren Nachbeobachtungszeit von Imatinib-behandelten Patienten ist der „Endpunkt mittleres Überleben“ noch nicht erreicht.
Stammzelltransplantation bei CML
Das Zehnjahresüberleben von mit Imatinib behandelten Patienten liegt deutlich höher als nach Stammzelltransplantation, wo die Überlebensraten historisch mit etwa 55 Prozent nach zehn Jahren angegeben werden (siehe die Tabelle der Risikoscores). Bei Stammzelltransplantation verstirbt ein erheblicher Teil der Patienten an den Folgen der intensiven Behandlung, an schweren Infektionen oder einer Graft-versus-host-Erkrankung. Auch die überlebenden Patienten nach Stammzelltransplantation haben häufig mit dem Problem einer chronischen graft-versus-host-Erkrankung zu kämpfen, so dass sie zwar kein Imatinib mehr einnehmen müssen, dafür aber andere Medikamente gegen die GvH-Erkrankung.
Heute ist es daher nicht mehr wie früher gerechtfertigt, CML-Patienten gleich nach Diagnosestellung einer Transplantation zuzuführen. Wohl aber kann es sinnvoll sein, bei einzelnen Hochrisikopatienten im Krankheitsverlauf eine Transplantation durchzuführen, wenn diese zum Beispiel kein Ansprechen auf die Behandlung mit Imatinib oder anderen Tyrosinkinase-Inhibitoren zeigen und sonst in gutem Allgemeinzustand sind. Unter Interferon-α-Therapie leben nach zehn Jahren noch 40 Prozent, von den Hochrisikogruppen nur 20 Prozent.
Komplette Heilung ohne Stammzelltransplantation?
Eine komplette Heilung im Sinne eines völligen Verschwindens des BCR-ABL-Onkogens war nach der lange Zeit vorherrschenden Expertenmeinung in der Regel weder durch die bisherigen Tyrosinkinase-Inhibitoren (Imatinib, Nilotinib, Dasatinib) noch durch Interferon-α zu erreichen. Es gibt Patienten, bei denen Imatinib oder Nilotinib so gut wirken, dass das BCR-ABL-Onkogen selbst mit den empfindlichsten Messmethoden nicht mehr nachweisbar ist. Hier ist im Einzelfall unklar, ob diese Patienten als geheilt gelten können, oder ob einfach die Messempfindlichkeit der Untersuchungsmethode nicht ausreicht, um die wenigen verbliebenen CML-Zellen nachzuweisen. Zurzeit wird deswegen in diesen Fällen nicht empfohlen, die Medikamente unkontrolliert wegzulassen, da ein Wiederauftreten der Erkrankung befürchtet wird.
Klinische Studien laufen, in denen unter anderem untersucht wird, ob man bei einer solchen oben beschriebenen BCR-ABL-Negativität mit immuntherapeutischen Ansätzen (zum Beispiel Interferon) eventuell die letzten verbliebenen CML-Stammzellen eliminieren und damit den Patienten komplett heilen kann.[32] Die schon weiter oben angesprochene deutsche TIGER-Therapiestudie, die im Jahr 2013 startete, untersucht diesen Therapieansatz. Geplant ist hier, Patienten primär mit Nilotinib (und zum Teil auch mit Interferon) zu behandeln. Bei Patienten, die eine über einen Zeitraum von mehr als 2 Jahren anhaltende komplette molekulare Remission erreichen, soll ein kompletter Auslassversuch gemacht werden – natürlich unter weiteren kontinuierlichen molekularen Kontrollen –, um zu sehen, ob die Patienten dauerhaft geheilt sind.[20]
Literatur
- Bubnoff, Nikolas von; Duyster, Justus: Chronische myeloische Leukämie: Therapie und Monitoring. In: Dtsch Arztebl Int. Nr. 107(7), 2010, S. 114–121 (Abstract).
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 376–449, hier: S. 424–426: Die chronisch-myeloische Leukämie (chronisch-leukämische Myelose).
Weblinks
- Informationen rund um die Erkrankung CML. CML-Info.com
- Onkologie, Hämatologie – Daten und Information. CML ONKODIN
- Informationen von Patienten für Patienten. Leukämie-Online
- Ratgeber Chronische myeloische Leukämie
Einzelnachweise
- Chronische myeloische Leukämie Leitlinie. Deutsche Gesellschaft für Hämatologie und Onkologie (DGHO), September 2013, abgerufen am 19. März 2016.
- Junia V. Melo, David J. Barnes: Chronic myeloid leukaemia as a model of disease evolution in human cancer. In: Nature Reviews Cancer. Band 7, 2007, S. 441–453, PMID 17522713 (nature.com [abgerufen am 8. August 2011]).
- SEER Stat Fact Sheets: Chronic Myeloid Leukemia. Surveillance, Epidemiology and End Results (SEER), abgerufen am 2. Juni 2011 (englisch, nach Daten 2004-2008).
- C. A. Schiffer: BCR-ABL Tyrosine kinase inihitors for chronic myelogenous leukemia. In: N Engl J Med. 2007; 357, S. 258–265. PMID 17634461
- Die Zahlenangabe für Deutschland beruht auf Schätzungen des Kompetenznetzes Akute und Chronische Leukämien weblink, die Zahlen für Österreich und die Schweiz sind proportional zu den deutschen Zahlen geschätzt
- A. Hochhaus: Chronische myeloische Leukämie (CML). In: S. Seeber, J. Schütte (Hrsg.): Therapiekonzepte Onkologie. Springer-Verlag, Heidelberg 2007, ISBN 978-3-540-28588-5, S. 293.
- D. G. Savage u. a.: Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period. In: Br J Haematol. 1997;96, S. 111–116. PMID 9012696.
- von altgriechisch χλωρός chlōrós „blass-/hellgrün, grüngelb“. Die Infiltrate erscheinen bei der Aufarbeitung durch den Pathologen leicht grünlich.
- European LeukemiaNet: CML recommendations
- R. Hehlmann, H. Heimpel, J. Hasford, H. J. Kolb, H. Pralle, D. K. Hossfeld, W. Queisser, H. Löffler, A. Hochhaus, B. Heinze u. a.: Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. In: Blood. 1994;84(12), S. 4064–4077. PMID 7994025.
- S. G. O’Brien, F. Guilhot, R. A. Larson, I. Gathmann, M. Baccarani, F. Cervantes, J. J. Cornelissen, T. Fischer, A. Hochhaus, T. Hughes, K. Lechner, J. L. Nielsen, P. Rousselot, J. Reiffers, G. Saglio, J. Shepherd, B. Simonsson, A. Gratwohl, J. M. Goldman, H. Kantarjian, K. Taylor, G. Verhoef, A. E. Bolton, R. Capdeville, B. J. Druker; IRIS Investigators: Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. In: N Engl J Med. 2003; 348, S. 994–1004. PMID 12637609
- FDA infopage Gleevec (Imatinib Mesylate), speziell Regulatory history including approval letters and reviews from Drugs@FDA
- Europäischer öffentlicher Beurteilungsbericht (EPAR): Glivec der Europäischen Arzneimittelagentur (pdf)
- M. Baccarani, J. Cortes, F. Pane, D. Niederwieser, G. Saglio, J. Apperley, F. Cervantes, M. Deininger, A. Gratwohl, F. Guilhot, A. Hochhaus, M. Horowitz, T. Hughes, H. Kantarjian, R. Larson, J. Radich, B. Simonsson, R. T. Silver, J. Goldman, R Hehlmann; European LeukemiaNet: Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009;27(35), S. 6041–6051. PMID 19884523
- U. Proetel, N. Pletsch, M. Lauseker, M. C. Müller, B. Hanfstein, S. W. Krause, L. Kalmanti, A. Schreiber, D. Heim, G. M. Baerlocher, W. K. Hofmann, E. Lange, H. Einsele, M. Wernli, S. Kremers, R. Schlag, L. Müller, M. Hänel, H. Link, B. Hertenstein, M. Pfirrman, A. Hochhaus, J. Hasford, R. Hehlmann, S. Saußele; German Chronic Myeloid Leukemia Study Group; Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK): Older patients with chronic myeloid leukemia (≥65 years) profit more from higher imatinib doses than younger patients: a subanalysis of the randomized CML-Study IV. In: Ann Hematol. 2014 Jul;93(7), S. 1167–1176. PMID 24658964.
- M. Baccarani, M. W. Deininger, G. Rosti, A. Hochhaus, S. Soverini, J. F. Apperley, F. Cervantes, R. E. Clark, J. E. Cortes, F. Guilhot, H. Hjorth-Hansen, T. P. Hughes, H. M. Kantarjian, D. W. Kim, R. A. Larson, J. H. Lipton, F. X. Mahon, G. Martinelli, J. Mayer, M. C. Müller, D. Niederwieser, F. Pane, J. P. Radich, P. Rousselot, G. Saglio, S. Saußele, C. Schiffer, R. Silver, B. Simonsson, J. L. Steegmann, J. M. Goldman, R. Hehlmann: European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. In: Blood. 2013;122(6), S. 872–884. doi:10.1182/blood-2013-05-501569. PMID 23803709.
- H. de Lavallade, J. F. Apperley, J. S. Khorashad, D. Milojkovic, A. G. Reid, M. Bua, R. Szydlo, E. Olavarria, J. Kaeda, J. M. Goldman, D. Marin: Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. In: J Clin Oncol. 2008 Jul 10;26(20), S. 3358–3363. PMID 18519952
- M. Talpaz u. a.: Dasatinib in Imatinib-Resistant Philadelphia Chromosome-Positive Leukemias. In: N Engl J Med. 2006;354, S. 2531–2541. PMID 16775234
- H. Kantarjian u. a.: Nilotinib in Imatinib-Resistant CML and Philadelphia Chromosome-positive ALL. In: N Engl J Med. 2006; 354, S. 2542–2551. PMID 17715389
- A. Hochhaus, T. Ernst, J, Ziermann, E. Eigendorff, P. La Rose: Chronische myeloische Leukämie. In: Onkologe. 2012; 18, S. 1108–1114. Springer-Verlag
- Ponatinib: a pan-BCR-ABL inhibitor. (Memento vom 26. November 2012 im Internet Archive)
- T. O’Hare, C. A. Eide, M. W. Deininger: Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. In: Blood. 2007 Oct 1;110(7), S. 2242–2249. PMID 17496200
- S. Branford, J. V. Melo, T. P. Hughes: Selecting optimal second-line tyrosine kinase inhibitor therapy for chronic myeloid leukemia patients after imatinib failure: does the BCR-ABL mutation status really matter? In: Blood. 2009 Dec 24;114(27), S. 5426–5435. PMID 19880502
- T. O’Hare, C. A. Eide, M. W. N. Deininger: Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. In: Blood. 2007;110(7), S. 2242–2249. PMID 17496200
- S. Redaelli, R. Piazza, R. Rostagno u. a.: Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. In: J Clin Oncol. 2009;27(3), S. 469–471. PMID 19075254
- S. Soverini, A. Hochhaus, F. E. Nicolini, F. Gruber, T. Lange, G. Saglio, F. Pane, M. C. Müller, T. Ernst, G. Rosti, K. Porkka, M. Baccarani, N. C. Cross, G. Martinelli: BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. In: Blood. 2011;118(5), S. 1208–1215. doi:10.1182/blood-2010-12-326405. PMID 21562040.
- Glivec 400 mg Filmtabletten. Abgerufen am 22. Januar 2022.
- „Primum non nocere“ und Profitstreben der pharmazeutischen Industrie - ein unauflösbarer Widerspruch in der Onkologie? In: Der Arzneimittelbrief Jahrgang 46, Nr. 5, Mai 2013.
- Experts in Chronic Myeloid Leukemia: The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. In: Blood. 2013;121(22), S. 4439–4442. PMID 23620577
- Glivec patent case in India. Abgerufen am 3. April 2014 (englisch, Darstellung des Falls aus Sicht der Fa. Novartis).
- CIVIL APPEAL Nos. 2706-2716 OF 2013. (Nicht mehr online verfügbar.) Supreme Court of India, 1. April 2013, archiviert vom Original am 7. April 2014; abgerufen am 3. April 2014 (englisch).
- A. Burchert, M. C. Müller, P. Kostrewa, P. Erben, T. Bostel, S. Liebler, R. Hehlmann, A. Neubauer, A. Hochhaus: Sustained molecular response with interferon alfa maintenance after induction therapy with imatinib plus interferon alfa in patients with chronic myeloid leukemia. In: J Clin Oncol. 2010;28(8), S. 1429–1435. PMID 20142590.