Chronische lymphatische Leukämie
Die chronische lymphatische Leukämie (englisch chronic lymphocytic leukemia, CLL), auch chronisch-lymphatische Leukämie genannt, ist ein niedrigmalignes, leukämisch verlaufendes B-Zell-Non-Hodgkin-Lymphom (B-NHL). Sie ist in der westlichen Welt die am häufigsten vorkommende Leukämieform und tritt vor allem im höheren Lebensalter auf. Das mediane Alter bei der Diagnosestellung liegt bei etwa 70 Jahren, weshalb die Erkrankung gelegentlich umgangssprachlich als Altersleukämie bezeichnet wird.
| Klassifikation nach ICD-10 | |
|---|---|
| C91.1 | Chronische lymphatische Leukämie ICD-O 9823/3 (CLL) ICD-O 9670/3 (B-SLL) |
| ICD-10 online (WHO-Version 2019) | |
Die WHO-Klassifikation der hämatologischen Erkrankungen unterscheidet neben der CLL noch eine Unterform, das kleinzellige B-Zell-Lymphom (englisch small lymphocytic lymphoma, B-SLL, oft auch bezeichnet als: SLL oder kleinzelliges lymphozytisches Lymphom[1]). Das kleinzellige B-Zell-Lymphom entspricht im Wesentlichen einer CLL, bei der die betroffenen B-Lymphozyten jedoch nicht primär im Knochenmark und Blut zu finden sind, sondern in den Lymphknoten. Es kann somit als nicht leukämisch verlaufende CLL bezeichnet werden.
Epidemiologie
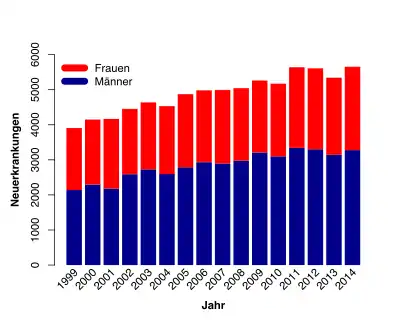
In Deutschland erkranken pro Jahr etwa 5.500 Menschen neu an CLL. Mit einem Anteil von 40 % an den neu diagnostizierten Leukämien ist die CLL damit die häufigste Leukämieform in Deutschland.[4] Die Statistiken zeigen in den letzten Jahren einen leichten Anstieg der Neuerkrankungen. Dieser Umstand beruht wahrscheinlich zum einen darauf, dass die Lebenserwartung der Menschen steigt[5], und zum anderen auf der verbesserten Diagnostik und der damit verbundenen Folge, dass mehr Patienten in einem frühen Stadium bei Routineuntersuchungen diagnostiziert werden.[6]
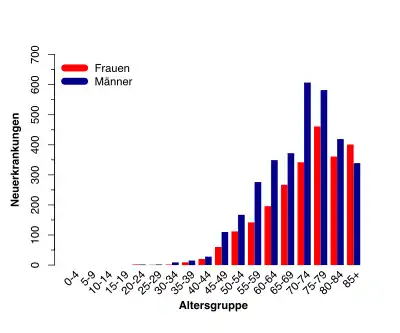
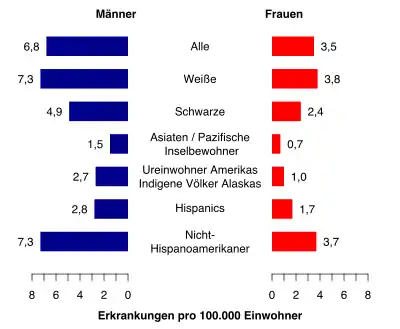
Die Erkrankungsrate entspricht ca. 6 Neuerkrankungen pro 100.000 Einwohner im Jahr.[7] Männer erkranken ungefähr im Verhältnis 1,4 : 1 etwas häufiger als Frauen an CLL. Das mediane Alter beträgt bei der Erstdiagnose 70 bis 75 Jahre.[8]
Die CLL zeigt einen deutlichen Bezug zu einer vorhandenen genetischen Veranlagung. Im asiatisch-pazifischen Raum gibt es nur wenige Neuerkrankungen. Die Erkrankungsraten steigen aus diesem Blickwinkel in Richtung der westlichen Länder stetig an.[9] Dass die Lebensumstände einen geringeren Einfluss haben, zeigt der Blick auf die Neuerkrankungen in den USA; diese spiegeln das Bild der steigenden Erkrankungsraten von den östlichen zu den westlichen Ländern anhand der multikulturellen Einwohner wider.[3]
Zudem weisen bis zu 10 % der Erkrankten in der Familiengeschichte eine Häufung an CLL auf. Das Risiko an CLL zu erkranken ist für direkte Nachkommen eines an CLL-Erkrankten im Vergleich zur restlichen Bevölkerung um einen Faktor 8,5 höher. Dieser Zusammenhang der genetischen Veranlagung zu den Erkrankungen ist im Moment noch nicht vollständig verstanden.[9]
Pathogenese
Bei der Erkrankung kommt es zu einer klonalen Vermehrung von reifen, kleinzelligen, aber funktionslosen B-Lymphozyten. Die genaue Ursache hierfür ist bisher weitgehend unbekannt. Es wird aber heute angenommen, dass genetische Veränderungen, die im Laufe des Lebens erworben wurden, entscheidende Auslöser der Krankheit sind.
Mehrere Metaanalysen zeigten einen quantitativen und statistischen Zusammenhang zwischen der Exposition von Herbiziden und einem erhöhten Risiko, an einem Non-Hodgkin-Lymphom zu erkranken. Dies wird allerdings kontrovers diskutiert und zum Teil unterschiedlich bewertet.[10][11][12][13]
Hinweise für eine infektiöse Ursache, z. B. durch Viren, gibt es bisher nicht.
Die molekulargenetische Analyse mittels Fluoreszenz-in-situ-Hybridisierung (FISH) zeigt in über 80 % genetische Veränderungen von Chromosomen. Die häufigste Veränderung ist eine Deletion auf Chromosom 13 (del(13q)). Weitere Veränderungen sind Deletionen von Chromosom 11 (del(11q)) und 17 (del(17p)) sowie eine Trisomie 12. Diese Chromosomenveränderungen haben prognostische Bedeutung. Patienten, die eine del(17p) aufweisen, haben typischerweise eine ungünstigere Prognose. Patienten mit einer del(13q) haben eine relativ günstige Prognose.
Überhaupt ist die CLL ein sehr heterogen verlaufendes Krankheitsbild. Es gibt Patienten, bei denen die Erkrankung einen sehr gutartigen Verlauf nimmt und über mehrere Jahre oder Jahrzehnte nicht behandelt werden muss. Bei Frauen wird im Vergleich zu Männern ein eher weniger aggressiver Verlauf und eine höhere Überlebenszeit beobachtet.[14] In etwa 1 bis 2 % der Fälle kommt es im Verlauf der Krankheit darüber hinaus zu dem ungewöhnlichen Phänomen einer teilweisen oder kompletten spontanen Remission.[15] Es gibt aber auch Patienten, bei denen die Erkrankung einen deutlich aggressiveren Verlauf zeigt. Diese Unterschiede im Krankheitsverlauf sind Folge verschiedener wechselseitiger Abhängigkeiten von genetischen Veränderungen und der Tumormikroumgebung bei einzelnen Patienten.
Diagnose
Ein Hinweis auf eine CLL wird heute meistens zufällig in einem frühen und beschwerdefreien Stadium bei einer routinemäßigen Blutbildkontrolle entdeckt. Das Untersuchungsergebnis weist in einem solchen Fall eine deutlich erhöhte Anzahl von Leukozyten auf. In der Regel werden daraufhin die nachfolgenden Blutuntersuchungen durchgeführt, um einen sicheren Nachweis der Diagnose zu erhalten:
- Differentialblutbild: Hierbei werden die weißen Blutkörperchen ausgezählt und entsprechend der Untergruppen eingeteilt. Eine für die klinische Diagnose der CLL notwendige Voraussetzung ist ein Nachweis von mindestens 5000 B-Lymphozyten/µl über ein Zeitraum von 3 Monaten im peripheren Blut.[6][16]
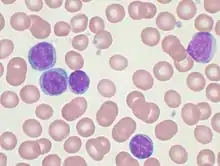
- Blutausstrich: Es wird ein Bluttropfen dünn auf einem Glasplättchen ausgestrichen, eingefärbt und mikroskopisch untersucht. Hierbei finden sich für die CLL typischen massenhaften kleinen und reifzelligen Lymphozyten, sowie häufig auch Gumprechtsche Kernschatten im Blutausstrich. Die Gumprechtschen Kernschatten entstehen durch das mechanische Zerquetschen der besonders empfindlichen CLL-Lymphozyten bei der Präparation des Blutausstrichs. Sie sind zwar häufig im Blutausstrich zu sehen, sind aber nicht kennzeichnend für eine CLL und finden sich ebenfalls bei anderen Non-Hodgkin-Lymphomen.[6][16]
- Immunphänotypisierung: Hierbei werden bestimmte Antigene auf der Zelloberfläche der Lymphozyten aus dem peripheren Blut mit unterschiedlichen fluoreszierenden Antikörpern eingefärbt und nach Art, Menge und Kombination mittels eines Durchflusszytometers ausgewertet. Die verschiedenen Lymphomarten weisen eine entsprechend charakteristische Zusammensetzung der Oberflächen-Antigene auf, die man als Immunphänotypen bezeichnet. Durch die Bestimmung des Immunphänotyps kann die CLL in der Regel sicher festgestellt und von anderen leukämisch verlaufenden Non-Hodgkin-Lymphomen unterschieden werden. Charakteristisch für CLL-Lymphozyten ist das gleichzeitige Vorhandensein des T-Zell-Antigens CD5 sowie der B-Zell-Oberflächenantigene CD19, CD20 und CD23, ebenso die geringe Ausprägung der Oberflächen-Immunglobuline sIgM und/oder sIgD und der Oberflächenantigene CD20 und CD79b. Bei der Immunphänotypisierung wird gleichzeitig auch die Monoklonalität der B-Zellen durch den Nachweis der sogenannten κ- oder λ-Leichtketten-Restriktion bestätigt.[6][16]
Weitere Untersuchungen dienen der Erkennung der Ausbreitung der CLL (Röntgen-Thoraxaufnahme, Ultraschalluntersuchung des Abdomens).
Differentialdiagnose
In der Regel kann die CLL durch Untersuchungen des Bluts schnell und eindeutig diagnostiziert werden, da in den meisten Fällen eine typische Morphologie und ein charakteristischer Immunphänotyp vorliegen. Wenn jedoch eine Zytopenie vorhanden ist oder wenn im peripheren Blut nur eine geringe Anzahl monoklonaler B-Lymphozyten nachweisbar ist, aber gleichzeitig eine Lymphadenopathie/Splenomegalie vorliegt oder monoklonale B-Lymphozyten mit einer CD5+ Expression ohne einen für die CLL typischen Phänotyp vorhanden sind, müssen weitere Untersuchungen durchgeführt werden, um die folgenden Diagnosen sicher auszuschließen:[17]
- Mantelzell-Lymphom
- B-Prolymphozytenleukämie
- Makroglobulinämie Waldenström
- Cold Agglutinin Disease
- Monoklonale B-Zelllymphozytose
- Richter-Transformation
Symptome
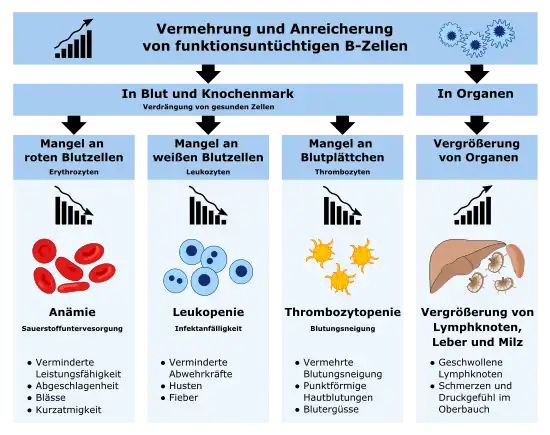
Häufig ist die Entdeckung der Krankheit ein Zufallsbefund während einer Blutuntersuchung im Rahmen der Diagnostik anderer Erkrankungen. Folgende Symptome und Befunde treten im Verlauf der Erkrankung auf:
- Lymphknotenschwellungen
- Milz/Lebervergrößerung
- Hauterscheinungen (30 Prozent der Fälle)
- Juckreiz
- Ekzeme
- Mykosen
- Herpes zoster
- Hautblutungen
- knotige Infiltrate
- blasse Haut und Schleimhaut
- Parotis/Ohrspeicheldrüsenschwellung (Mikulicz-Syndrom)
- Leukozytose mit Lymphozytose > 10.000 /µl
- im Knochenmark hoher Anteil reifer Lymphozyten
- Antikörpermangelsyndrom durch Verdrängung der B-Zellen
- Nachweis inkompletter Wärmeautoantikörper
- Autoimmunthrombozytopenie im Rahmen eines Evans-Syndrom
- verminderte Antikörper
- erhöhte IgM
- erhöhte Werte für Thymidinkinase und β2-Mikroglobulin
Stadieneinteilung und Prognosen
In der Praxis werden im Moment die Stadieneinteilungen nach Binet und Rai genutzt. Ersteres Einteilungsschema wird vorwiegend in Europa verwendet, letzteres in den USA. Beide Stadieneinteilungen haben sich aufgrund der einfachen und kostengünstigen Anwendbarkeit durchgesetzt. Vergrößerungen von Lymphknoten, Leber und Milz werden ausschließlich durch ertasten beurteilt, des Weiteren werden noch die Hämoglobin- und Thrombozytenwerte aus dem Blutbild verwendet.
Bei Binet wird anhand der Anzahl der betroffenen Lymphknoten-Regionen und dem Vorliegen einer Anämie beziehungsweise einer Thrombopenie in drei Stadien mit unterschiedlichen Prognosen unterteilt. Die dabei angegebene mediane Überlebenszeit beruht auf historischen Daten wie sie in den 1970er und 1980er Jahren zur Verfügung standen; es ist davon auszugehen, dass die Prognosen heute aufgrund neuer Behandlungsmethoden weitaus positiver sind.
| Stadium | Anzahl der betroffenen Lymphknotenregionen* |
Hämoglobin [g/dl] |
Thrombozyten [G/l] |
Medianes Überleben [Jahre] |
|---|---|---|---|---|
| A | < 3 | ≥ 10,0 | ≥ 100,0 | > 10 |
| B | ≥ 3 | ≥ 10,0 | ≥ 100,0 | 5-7 |
| C | irrelevant | < 10,0 | < 100 | 2,5-3 |
Das in den USA gebräuchliche Einteilungssystem nach Rai unterscheidet im Gegensatz zu Binet fünf Stadien und es wird ein Hämoglobin-Wert von < 11,0 g/dl anstatt von < 10,0 g/dl als Grenzwert für das Vorliegen einer Anämie verwendet. Das System von Rai wurde im Verlauf der Anwendung allerdings ebenfalls auf drei Gruppen zusammengefasst, die dem Einteilungssystem von Binet entsprechen. Dabei wurde das Stadium 0 als niedriges Risiko, das Stadium I und II als mittleres Risiko und das Stadium III und IV als hohes Risiko klassifiziert.
| Stadium |
Lymphadenopathie |
Hepato- oder Splenomegalie |
Hämoglobin [g/dl] |
Thrombozyten [G/l] |
Medianes Überleben [Jahre] |
|---|---|---|---|---|---|
| Niedriges Risiko | |||||
| 0 | keine | keine | ≥ 11,0 | ≥ 100,0 | > 10 |
| Mittleres Risiko | |||||
| I | ≥ 1 | keine | ≥ 11,0 | ≥ 100,0 | 5-7 |
| II | irrelevant | ≥ 1 | ≥ 11,0 | ≥ 100,0 | |
| Hohes Risiko | |||||
| III | irrelevant | irrelevant | < 11,0 | ≥ 100,0 | 2,5 - 3 |
| IV | irrelevant | irrelevant | irrelevant | < 100,0 | |
Der Krankheitsverlauf variiert allerdings stark zwischen den Patienten eines Stadiums. Damit eine bessere individuelle Prognose erstellt werden kann, wurde ergänzend ein Prognoseindex erarbeitet. Bei dem Index werden die verschiedenen heute bekannten unabhängigen Einflussfaktoren für einen günstigen oder ungünstigen Verlauf mit einer entsprechenden Gewichtung berücksichtigt. Zu diesen Einflussfaktoren gehören die beiden genetischen Faktoren TP53 Status und der IGHV-Mutationsstatus, so wie der Serum-β2-Mikroglobulin-Spiegel als biochemischer Faktor und die beiden klinischen Faktoren Krankheitsstadium und Alter des Patienten.
| Unabhängiger Prognosefaktor | Ausprägung | Punktwert |
|---|---|---|
| TP53 Status | Deletiert oder mutiert | 4 |
| IGHV-Mutationsstatus | unmutiert | 2 |
| Serum-β2-Mikroglobulin | > 3,5 mg/L | 2 |
| Krankheitsstadium | Binet B-C oder Rai I-IV | 1 |
| Alter | > 65 | 1 |
Die den Faktoren zugewiesenen Punktzahlen werden zu einer Gesamtpunktwert aufaddiert, mit dessen Hilfe zwischen vier Risikogruppe unterschieden werden kann. Jede dieser Risikogruppe unterscheidet sich in dem statistischen ermittelten Gesamtüberleben nach fünf und zehn Jahren.
| Risikogruppe | Gesamtpunktwert | Gesamtüberleben nach 5 Jahren [%] |
Gesamtüberleben nach 10 Jahren [%] |
Median Gesamtüberlebenszeit [Monate] |
|---|---|---|---|---|
| Niedriges Risiko | 0-1 | 93,2 | 79,0 | - |
| Mittleres Risiko | 2-3 | 79,3 | 39,2 | 105 |
| Hohes Risiko | 4-6 | 63,3 | 21,9 | 75 |
| Sehr hohes Risiko | 7-10 | 23,3 | 3,5 | 29 |
Therapie
CLL ist durch konventionelle Chemotherapie oder durch Antikörpertherapie nach derzeitigem Kenntnisstand nicht heilbar. Durch Knochenmarkstransplantation ist eine Heilung prinzipiell möglich. Die Therapie hängt vom jeweiligen Stadium der Erkrankung ab. Durch die rasante Entwicklung neuer molekularbiologischer Diagnoseverfahren, neuer Medikamente und Behandlungsschemata ändert sich die empfohlene Therapie schnell. Die DKG empfiehlt, allen Patienten die Teilnahme an einer klinischen Studie anzubieten.
Die klinische Einteilung nach Binet unterscheidet drei Stadien. In frühen Stadien (Binet Stadium A und B) wird in der Regel noch nicht behandelt, es sei denn, die Erkrankung verursacht Beschwerden oder schreitet sehr schnell voran. Diese Beschwerden können sein:
- Milzvergrößerung mit Symptomen
- Beschwerden durch wachsende Lymphknoten
- schwere, die Lebensqualität beeinträchtigende Allgemeinsymptome (Nachtschweiß, wiederholte Infekte, Fieber, Gewichtsverlust)
Eine Behandlung ist darüber hinaus in der Regel angezeigt ab dem Stadium Binet C (schwere Anämie oder Thrombozytopenie).
Die Therapie hängt vom Zustand des Patienten und den genetischen Mutationen der betroffenen B-Lymphozyten ab.[22] Als Erstlinientherapie von Patienten in gutem Zustand ("fit") wird eine Kombination der Chemotherapeutika Fludarabin und Cyclophosphamid mit dem Antikörper Rituximab empfohlen. Eine Alternative hierzu ist die Kombination von Bendamustin mit Rituximab (Bendamustin ist seit 2010 für die Behandlung der CLL zugelassen). Für Patienten in weniger gutem Zustand ("unfit") wird eine Kombination aus Chlorambucil bzw. Bendamustin mit dem Antikörper Rituximab empfohlen. Dieser kann durch die neueren Antikörper Ofatumumab oder Obinutuzumab ersetzt werden, die das gleiche Zielmolekül wie Rituximab erkennen (CD20). Obinutuzumab hat in einer Studie eine höhere Ansprechrate im Vergleich zu Rituximab gezeigt.[23] Bei gebrechlichen Patienten wird eine Supportivtherapie empfohlen.
Auch die Zweitlinientherapie, d. h. die Therapie für Patienten, die nach einer ersten Therapie einen Rückfall erleiden, hängt vom Zustand des Patienten und den genetischen Mutationen der Tumorzellen ab.[22] Falls zwischen der Primärerkrankung und dem Rückfall mehr als 2–3 Jahre vergangen sind, kann man noch einmal die Primärtherapie durchführen, wie sie im letzten Absatz beschrieben ist. Ansonsten wird bei den meisten Patienten entweder der Bruton-Tyrosinkinase-Inhibitor Ibrutinib[24] oder eine Kombination des PI3-Kinase-Inhibitors Idelalisib[25] mit Rituximab oder der Anti-CD52-Antikörper Alemtuzumab empfohlen. Zu Idelalisib wurde im März 2016 ein Rote-Hand-Brief (u. a. nicht als Erstlinientherapie!) veröffentlicht.[26] Gebrechliche Patienten sollten Supportivtherapie erhalten. Bei der Zweitlinientherapie können auch andere Cytostatika zum Einsatz kommen. Im Idealfall sollte die Behandlung im Rahmen einer klinischen Studie erfolgen.
Des Weiteren kommt eine Knochenmark- oder Stammzelltransplantation in Betracht. Allerdings sind die allogenen Transplantationsstrategien bei der CLL mit hohen therapiebedingten Sterblichkeitsraten verbunden und kommen nur bei ausgewählten Patienten zur Anwendung. Inzwischen werden Stammzelltransplantationen mit reduzierter Konditionierung auch bei älteren Patienten angewendet.[27] Größere Lymphome kann man mit einer Strahlentherapie behandeln.
Bei einer Stammzelltransplantation kann es zusätzlich noch zu einer Graft-versus-Host Reaktion kommen. Es wurde evaluiert, ob mesenchymale Stromazellen zur Prophylaxe und Therapie verwendet werden können.[28]
Die häufig belastende Behandlung der CLL kann durch körperliche Betätigung ergänzt werden, um das Wohlbefinden zu verbessern.[29]
Literatur
- Michael Hallek, Barbara Eichhorst, Daniel Catovsky (Hrsg.): Chronic Lymphocytic Leukemia (= Hematologic Malignancies). 1. Auflage. Springer Nature Switzerland, Cham 2019, ISBN 978-3-03011391-9 (englisch).
- Michael Hallek, Barbara Eichhorst (Hrsg.): Chronische lymphatische Leukämie (= UNI-MED SCIENCE). 5. Auflage. UNI-MED, Bremen 2014, ISBN 978-3-8374-2263-4.
- Hermann Delbrück: Chronische Leukämien. Rat und Hilfe für Betroffene und Angehörige (= Rat & Hilfe). 3. Auflage. Kohlhammer, Stuttgart 2008, ISBN 978-3-17-020470-6.
- Guy B. Faguet (Hrsg.): Chronic Lymphocytic Leukemia. Molecular Genetics, Biology, Diagnosis, and Management (= Contemporary Hematology). Humana Press, Totowa, New Jersey 2004, ISBN 1-58829-099-9 (englisch).
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/ Göttingen/ Heidelberg 1955. (2. Auflage ebenda 1961, S. 376–449, hier: S. 426–428: Die chronisch-lymphatische Leukämie (chronisch-leukämische Lymphadenose).)
Weblinks
Einzelnachweise
- Selbsthilfevereinigung zur Unterstützung erwachsener Leukämie- und Lymphompatienten (SELP e.V.) Abgerufen am 29. Juli 2021.
- Datenbankabfrage - Zentrum für Krebsdaten. Robert Koch-Institut, abgerufen am 26. Oktober 2019.
- Cancer Stat Facts: Leukemia - Chronic Lymphocytic Leukemia (CLL). National Cancer Institute, abgerufen am 11. November 2019 (eng).
- Zentrum für Krebsregisterdaten und Gesellschaft der epidemiologischen Krebsregister in Deutschland e.V.: Krebs in Deutschland für 2013/2014. (PDF) Robert Koch-Institut, 2017, S. 128 ff., abgerufen am 1. November 2019.
- Hermann Delbrück: Chronische Leukämien. Rat und Hilfe für Betroffene und Angehörige (= Rat & Hilfe). 3. Auflage. Kohlhammer, Stuttgart 2008, ISBN 978-3-17-020470-6, S. 34.
- Michael Hallek, Barbara Eichhorst (Hrsg.): Chronische lymphatische Leukämie (= UNI-MED SCIENCE). 5. Auflage. UNI-MED, Bremen 2014, ISBN 978-3-8374-2263-4, Diagnostik.
- Was ist chronische lymphatische Leukämie (CLL). Häufigkeit und Erkrankungsalter. Deutsche CLL Studiengruppe (DCLLSG), abgerufen am 15. November 2019.
- Kurt Possinger, Anne Regierer (Hrsg.): Facharzt Hämatologie und Onkologie. Elsevier, München, ISBN 978-3-437-23770-6.
- Michael Hallek, Barbara Eichhorst, Daniel Catovsky (Hrsg.): Chronic Lymphocytic Leukemia (= Hematologic Malignancies). 1. Auflage. Springer Nature Switzerland, Cham 2019, ISBN 978-3-03011391-9, Chronic Lymphocytic Leukemia: Who, How, and Where? (englisch).
- Luoping Zhang, Iemaan Rana, Rachel M. Shaffer, Emanuela Taioli, Lianne Sheppard: Exposure to glyphosate-based herbicides and risk for non-Hodgkin lymphoma: A meta-analysis and supporting evidence. In: Mutation Research Reviews in Mutation Research. Band 781. Elsevier B.V., 10. Februar 2019, S. 186–206, doi:10.1016/j.mrrev.2019.02.001 (englisch).
- Ellen T. Chang, Elizabeth Delzell: Systematic review and meta-analysis of glyphosate exposure and risk of lymphohematopoietic cancers. In: Journal of Environmental Science and Health, Part B. Band 51, Nr. 6. Taylor & Francis, 25. März 2016, S. 402–428, doi:10.1080/03601234.2016.1142748 (englisch).
- Leah Schinasi, Maria E. Leon: Non-Hodgkin Lymphoma and Occupational Exposure to Agricultural Pesticide Chemical Groups and Active Ingredients: A Systematic Review and Meta-Analysis. In: International Journal of Environmental Research and Public Health. Band 11, Nr. 4. Springer Science+Business Media S.A., 23. April 2014, S. 4440–4527, doi:10.3390/ijerph110404449 (englisch).
- Some Organophosphate Insecticides and Herbicides. In: IARC Working Group (Hrsg.): IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Band 112, 10. März 2015 (englisch, iarc.fr [PDF; abgerufen am 22. Juni 2020]).
- Michael Hallek, Barbara Eichhorst, Daniel Catovsky (Hrsg.): Chronic Lymphocytic Leukemia (= Hematologic Malignancies). 1. Auflage. Springer Nature Switzerland, Cham 2019, ISBN 978-3-03011391-9, The Clinical Presentation of CLL (englisch).
- Marwan Kwok, Ceri Oldreive, Andy C. Rawstron, Anshita Goel, Grigorios Papatzikas, Rhiannon E. Jones, Samantha Drennan, Angelo Agathanggelou, Archana Sharma-Oates, Paul Evans, Edward Smith, Surita Dalal, Jingwen Mao, Robert Hollows, Naheema Gordon, Mayumi Hamada, Nicholas J. Davies, Helen Parry, Andrew D. Beggs, Thalha Munir, Paul Moreton, Shankara Paneesha, Guy Pratt, A. Malcolm R. Taylor, Francesco Forconi, Duncan M. Baird, Jean-Baptiste Cazier, Paul Moss, Peter Hillmen, Tatjana Stankovic: Integrative analysis of spontaneous CLL regression highlights genetic and microenvironmental interdependency in CLL. In: Blood. Band 135, Nr. 6, 6. Februar 2020, S. 411–428, doi:10.1182/blood.2019001262 (englisch).
- Michael Hallek, Bruce D. Cheson, Daniel Catovsky, Federico Caligaris-Cappio, Guillermo Dighiero, Hartmut Döhner, Peter Hillmen, Michael Keating, Emili Montserrat, Nicholas Chiorazzi, Stephan Stilgenbauer, Kanti R. Rai, John C. Byrd, Barbara Eichhorst, Susan O’Brien, Tadeusz Robak, John F. Seymour, Thomas J. Kipps: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. In: Blood. Band 131, Nr. 25, 21. Juni 2018, doi:10.1182/blood-2017-09-806398 (englisch).
- Michael Hallek, Barbara Eichhorst, Daniel Catovsky (Hrsg.): Chronic Lymphocytic Leukemia (= Hematologic Malignancies). 1. Auflage. Springer Nature Switzerland, Cham 2019, ISBN 978-3-03011391-9, Laboratory Diagnosis of Chronic Lymphocytic Leukaemia (englisch).
- J. L. Binet: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. In: Cancer. Band 48, Nr. 1. Wiley-Blackwell, 1. Juli 1981 (englisch, wiley.com [PDF; abgerufen am 12. Oktober 2019]).
- S3-Leitlinie zur Diagnostik, Therapie und Nachsorge für Patienten mit einer Chronischen lymphatischen Leukämie (CLL). (PDF) Leitlinienprogramm Onkologie der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V., Deutsche Krebsgesellschaft e.V. (DKG) und Deutsche Krebshilfe (DKH), März 2018, S. 33-35, abgerufen am 18. Oktober 2019.
- The International CLL-IPI working group: An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. In: The Lancet Oncology. Band 17, Nr. 6. Elsevier, Juni 2016, ISSN 1474-5488, S. 779–790, doi:10.1016/S1470-2045(16)30029-8, PMID 27185642 (englisch).
- Michael Hallek, Barbara Eichhorst, Daniel Catovsky (Hrsg.): Chronic Lymphocytic Leukemia (= Hematologic Malignancies). 1. Auflage. Springer Nature Switzerland, Cham 2019, ISBN 978-3-03011391-9, Prognostic Markers (englisch).
- Federführende Fachgesellschaft: Deutsche Gesellschaft für Hämatologie und medizinische Onkologie (DGHO): S3-Leitlinie zur Diagnostik, Therapie und Nachsorge für Patienten mit einer chronischen lymphatischen Leukämie (CLL) Langversion 1.0 – März 2018 AWMF-Registernummer: 018-032OL. (PDF) Leitlinienprogramm Onkologie der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF), Deutschen Krebsgesellschaft e.V. (DKG) und Deutschen Krebshilfe (DKH)., 2018, abgerufen am 11. Juni 2019.
- Valentin Goede, Kirsten Fischer, Raymonde Busch, Anja Engelke, Barbara Eichhorst, Clemens Wendtner, Tatiana Chagorova, Javier de la Serna, Marie-Sarah Dilhuydy, Thomas Illmer, Stephen Opat, Carolyn J. Owen, Olga Samoylova, Karl-Anton Kreuzer, Stephan Stilgenbauer, Hartmut Döhner, Anton W. Langerak, Matthias Ritgen, Michael Kneba, Elina Asikanius, Kathryn Humphrey, Michael Wenger, Michael Hallek: Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. In: New England Journal of Medicine. Band 370, 2014, S. 1101–1110, doi:10.1056/NEJMoa1313984.
- Ibrutinib: Neuer Kinasehemmer bei Blutkrebs. In: Pharmazeutische Zeitung. 24. September 2014 (pharmazeutische-zeitung.de [abgerufen am 5. September 2015]).
- Leukämie und Lymphom: Neues Medikament zugelassen. In: Pharmazeutische Zeitung. 21. Oktober 2014 (pharmazeutische-zeitung.de [abgerufen am 5. September 2015]).
- Einschränkungen für die Anwendung von Idelalisib zur Behandlung der CLL, RHB Gilead vom 23. März 2016, abgerufen am 24. März 2016.
- IDW-Online 20. September 2011.
- Sheila A Fisher, Antony Cutler, Carolyn Doree, Susan J Brunskill, Simon J Stanworth: Mesenchymal stromal cells as treatment or prophylaxis for acute or chronic graft-versus-host disease in haematopoietic stem cell transplant (HSCT) recipients with a haematological condition. In: Cochrane Database of Systematic Reviews. 30. Januar 2019, doi:10.1002/14651858.CD009768.pub2 (wiley.com [abgerufen am 25. August 2020]).
- Linus Knips, Nils Bergenthal, Fiona Streckmann, Ina Monsef, Thomas Elter: Aerobic physical exercise for adult patients with haematological malignancies. In: Cochrane Database of Systematic Reviews. 31. Januar 2019, doi:10.1002/14651858.CD009075.pub3 (wiley.com [abgerufen am 25. August 2020]).