Organokatalyse
Unter Organokatalyse versteht man in der Organischen Chemie die Katalyse organischer Reaktionen mit Hilfe kleiner, metallfreier organischer Moleküle, die aus den Atomen Kohlenstoff, Wasserstoff, Sauerstoff, Stickstoff, Schwefel und Phosphor aufgebaut sind. Der Begriff wurde durch den deutschen Chemiker Wolfgang Langenbeck geprägt.[1] Im Jahr 2021 wurde den Chemikern Benjamin List und David MacMillan für ihre Forschung zur (asymmetrischen) Organokatalyse der Nobelpreis für Chemie zuerkannt.[2]
Geschichte

Als Beginn der Organokatalyse gilt die 1832 von Justus von Liebig und Friedrich Wöhler entdeckte Benzoin-Addition unter Cyanid-Katalyse zu aromatischen α-Hydroxyketonen (Benzoin).[3]
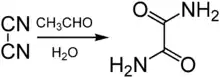
Im Jahr 1859 entdeckte Justus von Liebig auch die Oxamid-Synthese aus Dicyan und Wasser in Gegenwart von Acetaldehyd.[4] Liebig identifizierte Acetaldehyd als Katalysator für die Reaktion und erkannte in dessen Wirkung Parallelen zu den Fermenten (Enzymen).
Die erste asymmetrische organokatalytische Reaktion wurde 1912 von Georg Bredig und P. S. Fiske publiziert. So wurde die Cyanhydrinreaktion mit Benzaldehyd zu Mandelonitril mit Alkaloiden katalysiert. Die erreichten Enantiomerenüberschüsse lagen um 10 %.[5]
Jahrzehnte später konnten erstmals beachtliche Stereoselektivitäten in einer organokatalytischen Reaktion erzielt werden. Als Katalysator wurde die Aminosäure Prolin [(S)- oder (R)-Prolin] in einer Robinson-Anellierung verwendet, die zum Wieland-Miescher-Keton führt. Diese Reaktion wird heute nach ihren Entdeckern Hajos-Parrish-Eder-Sauer-Wiechert-Reaktion genannt und besaß für die Totalsynthesen von Steroiden große Bedeutung.[6][7][8]
Die ersten Umpolungskatalysatoren mit denen diese Reaktion auch mit aliphatischen Aldehyden durchgeführt werden konnte, wurden 1991 von Forschern der BASF entwickelt. Dabei werden Thiazolium-Katalysatoren verwendet.[9] Weitere Katalysatoren mit ähnlicher Funktion basieren auf Triazoliumyliden[10][11] und Perimidiniumyliden.[12]
| Katalytische Umpolung | ||
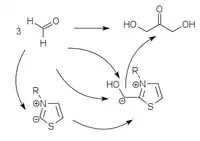 |
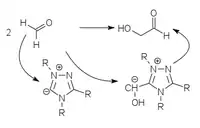 |
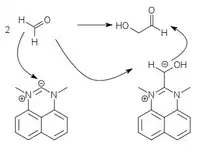 |
| Thiazolium-Katalyse | Triazolium-Katalyse | Perimidinium-Katalyse |
Durch das Houk-Modell wurde das erste Mal ein schlüssiger Mechanismus für die metallfreie Enamin-Aldolreaktion analog zum Zimmerman-Traxler-Modell vorgeschlagen.[13] Gekreuzte direkte Aldolreaktionen wurden unabhängig von Benjamin List[14], Carlos Barbas[15], Masakatsu Shibasaki[16] und Barry Trost[17] entwickelt. Über die erste organokatalytische gekreuzte Aldolreaktion von Aldehyden berichtete 2002 David MacMillan.[18]
Reaktionsmechanismus
Der Katalysator kann im Katalysezyklus kovalent an das Substratmolekül gebunden sein; in diesem Falle sind relativ hohe Konzentrationen des Organokatalysators erforderlich. Auch sind katalytische, nicht-kovalente Wechselwirkungen etwa über Wasserstoffbrückenbindungen möglich, die nur geringe Mengen des Katalysators erfordern.
Kovalenter Mechanismus
Das Prinzip der meisten organokatalytischen Verfahren besteht darin, dass der Katalysator zuerst mit einem Reaktionspartner unter Ausbildung einer (reversiblen) kovalenten Bindung reagiert. In der prolinkatalytischen Aldolreaktion kondensiert (S)-Prolin zunächst an das eingesetzte Keton. Das entstandene Iminiumion tautomerisiert dann zum Enamin, das im nächsten Schritt nukleophil am eingesetzten Aldehyd angreift. Durch anschließende Hydrolyse wird das Produkt freigesetzt und (S)-Prolin zurückgebildet.
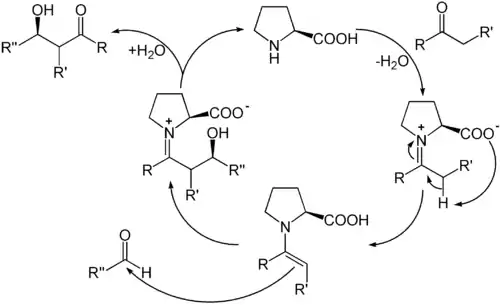
In der Reaktion wird die Stereoinformation durch das (chirale) (S)-Prolin übertragen. Die Carboxygruppe des (S)-Prolins aktiviert durch Ausbildung einer Wasserstoffbrückenbindung auch den Aldehyd. Die Reaktion verläuft über einen sechsgliedrigen sesselartigen Übergangszustand ähnlich dem Zimmerman-Traxler-Modell für Lithiumenolate. Der Substituent des Aldehyds liegt hierbei in der pseudo-äquatorialen Ebene.
Der Verlauf der Reaktion über einen sesselartigen Übergangszustand wurde zuerst durch quantenchemische Berechnungen von Houk postuliert[13] und später von List experimentell durch Sauerstoffmarkierung bewiesen.[19] Wenn statt (S)-Prolin die nichtproteinogene Aminosäure (R)-Prolin eingesetzt wird, entsteht mit gleicher Stereoselektivität das enantiomere Aldol.
Nicht-kovalente Organokatalyse
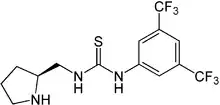
Bei der nicht-kovalenten Organokatalyse werden keine kovalenten Bindungen zum Katalysator ausgebildet. Sie basiert auf schwachen, gerichteten Wechselwirkungen zwischen einem Organokatalysator und dem zu aktivierenden Substrat. Nach diesem Prinzip reagieren auch viele Enzyme, die auch als Vorbild für die Entwicklung nicht-kovalenter Organokatalysatoren dienen. Als neutrale Wasserstoffbrückendonoren werden beispielsweise Derivate des Harnstoffs oder Thioharnstoffs eingesetzt.[20][21] Als günstig haben sich hierbei Katalysatoren erwiesen, die elektronenarm sowie von starrer Struktur sind und einen Phenylring besitzen, der in 3-, 4- und/oder 5-Position elektronenziehende, nicht koordinierende Substituenten tragen.
Vorteile von Thioharnstoffderivaten (vor allem gegenüber traditionellen, metall-haltigen Lewis-Säure-Katalysatoren):
- nicht-kovalente Bindung am Substrat und somit geringe Produktinhibition
- niedrige Katalysatorbeladung (bis 0,001 mol%), hohe TOF-Werte
- einfache und günstige Synthese und strukturelle Modifikation
- Anbindung an die Festphase; somit Rückgewinnung möglich
- nicht luft- oder wasserempfindlich, keine Inertgasatmosphäre nötig, unproblematische Handhabung
- ermöglichen Katalyse unter nahezu neutralen Bedingungen, Toleranz säurelabiler Substrate
- metall-frei, nicht toxisch wie viele metallhaltigen Lewis-Säure-Katalysatoren
- umweltverträglich („Grüne Chemie“)
Reaktionen
Für folgende Reaktionen gibt es bereits wirksame Organokatalysatoren:
- Aldolreaktionen (z. B. Hajos-Parrish-Eder-Sauer-Wiechert-Reaktion)
- Michael-Additionen
- Mannich-Reaktionen
- α-Aminierungen
- Diels-Alder- und Hetero-Diels-Alder-Reaktionen
- Knoevenagel-Kondensationen
- Wittig-Umlagerungen
- Fluorierungen
- Reduktionen
- asymmetrische Stetter-Reaktionen
- Nitroaldol-Reaktion
- Shi-Epoxidierung
- Baylis-Hillman-Reaktion
- Ketonreduktion
Von Naturstoffen abgeleitete Katalysatoren
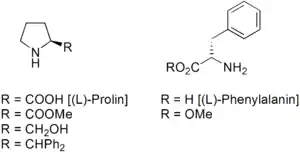
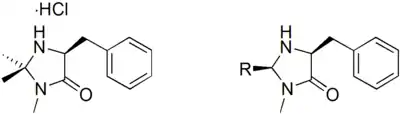
Häufig wurden und werden von der Aminosäuren (S)-Prolin abgeleitete Katalysatoren verwendet.[22][23] Auch vom (S)-Phenylalanin abgeleitete Katalysatoren werden oft angewandt.
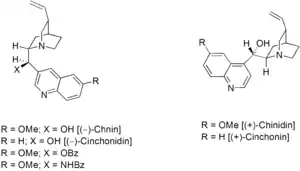
Von den Cinchona(China)-Alkaloiden leiten sich einige in organokatalytischen Reaktionen verwendete Katalysatoren ab:
Auch von Weinsäure abgeleitete Katalysatoren, zum Beispiel TADDOLE werden in organokatalytischen Reaktionen verwendet:

Einzelnachweise
- W. Langenbeck: Die organischen Katalysatoren und ihre Beziehungen zu den Fermenten, Springer, Berlin 1949.
- The Nobel Prize in Chemistry 2021. Abgerufen am 6. Oktober 2021 (amerikanisches Englisch).
- F. Wöhler und J. Liebig, Ann. Pharm. 1832, 3, 249.
- J. v. Liebig: Ueber die Bildung des Oxamids aus Cyan, in: Liebigs Ann. 1860, 113, 246–247; doi:10.1002/jlac.18601130213.
- G. Bredig, P. S. Fiske, Biochem Z 1912, 46, 7.
- U. Eder, G. Sauer, R. Wiechert: Neuartige asymmetrische Cyclisierung zu optisch aktiven Steroid-CD-Teilstücken, in: Angew. Chem. 1971, 10, 492–493; doi:10.1002/ange.19710831307.
- Z. G. Hajos, D. R. Parrish: Asymmetric Synthesis of Optically Active Polycyclic Organic Compunds. Deutsches Patent DE 2102623, 29. Juli 1971.
- Z. G. Hajos, D. R. Parrish: Asymmetric synthesis of bicyclic intermediates of natural product chemistry, in: J. Org. Chem. 1974, 39, 1615; doi:10.1021/jo00925a003.
- Patent DE000004122669A1: Verfahren zur Herstellung von Dihydroxyaceton. Veröffentlicht am 9. Juli 1991, Anmelder: BASF AG, 67063 Ludwigshafen, DE.
- Patent DE000004212264A1: Verfahren zur katalytischen Herstellung von Kondensationsprodukten des Formaldehyds. Veröffentlicht am 11. April 1992, Anmelder: BASF AG, 67063 Ludwigshafen, DE.
- Patent DE000004230466A1: Verfahren zur katalytischen Herstellung von Kondensationsprodukten des Formaldehyds. Veröffentlicht am 11. September 1992, Anmelder: BASF AG, 67063 Ludwigshafen, DE.
- Patent DE000019536403A1: Perimidiniumsalze, ihre Herstellung und Verwendung. Veröffentlicht am 29. Mai 1995, Anmelder: BASF AG, 67063 Ludwigshafen, DE.
- K. N. Houk, S. Bahmanyar: The Origin of Stereoselectivity in Proline-Catalyzed Intramolecular Aldol Reactions. In: J. Am. Chem. Soc. 2001, 123, 12911–12912, doi:10.1021/ja011714s.
S. Bahmanyar, K. N. Houk: Transition States of Amine-Catalyzed Aldol Reactions Involving Enamine Intermediates: Theoretical Studies of Mechanism, Reactivity, and Stereoselectivity. In: J. Am. Chem. Soc. 2001, 123, 11273–11283, doi:10.1021/ja011403h. - B. List, R. A. Lerner, C. F. Barbas, III., Proline-Catalyzed Direct Asymmetric Aldol Reactions in: J. Am. Chem. Soc. 2000, 122, 2395–2396, doi:10.1021/ja994280y.
- S. Kandasamy, W. Notz, T. Bui, C. F. Barbas, III., Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon-Carbon Bond-Forming Reactions in: J. Am. Chem. Soc. 2001, 123, 5260–5267, doi:10.1021/ja010037z.
- Y. M. A. Yamada, N. Yoshikawa, H. Sasai, M. Shibasaki: Direkte katalytische asymmetrische Aldolreaktionen von Aldehyden mit nicht modifizierten Ketonen in: Angew. Chem. 1997, 109, 1842–1944; doi:10.1002/ange.19971091716.
- B. M. Trost, H. Ito, A Direct Catalytic Enantioselective Aldol Reaction via a Novel Catalyst Design in: J. Am. Chem. Soc. 2000, 122, 12003–12004, doi:10.1021/ja003033n.
- A. B. Northrup, D. W. C. MacMillan, The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes in: J. Am. Chem. Soc. 2002, 124, 6798-6799, doi:10.1021/ja0262378.
- Linh Hoang, K. N. Houk, S. Bahmanyar, B. List: Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions. In: J. Am. Chem. Soc. 2003, 125, 16–17.
- M. S. Taylor, E. N. Jacobsen: Asymmetric Catalysis by Chiral Hydrogen-Bond Donors, in: Angew. Chem. Int. Ed. 2006, 45, 1520–1543; doi:10.1002/anie.200503132.
- S. J. Connon: Organocatalysis Mediated by (Thio)urea Derivatives, in: Chem. Eur. J. 2006, 12, 5418 –5427; doi:10.1002/chem.200501076.
- D. Seebach, A. K. Beck, D. M. Badine, M. Limbach, A. Eschenmoser, A. M. Treasurywala, R. Hobi, W. Prikoszovich, B. Linder: Are Oxazolidinones really unproductive, parasitic species in proline catalysis? Thoughts and experiments pointing to an alternative view, in: Helv. Chim. Acta 2007, 90, 425–471.
- S. Mukherjee, J. W. Yang, S. Hoffmann, B. List: Asymmetric enamine catalysis, in: Chem. Rev. 2007, 107, 5471–5569.
- K. A. Ahrendt, C. J. Borths, D. W. C. MacMillan, New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction in: J. Am. Chem. Soc. 2000, 122, 4243–4244, doi:10.1021/ja000092s.
- N. A. Paras, D. W. C. MacMillan, New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel-Crafts Alkylation in: J. Am. Chem. Soc. 2001, 123, 4370–4371, doi:10.1021/ja015717g.
- A. B. Northrup, D. W. C. MacMillan, The First General Enantioselective Catalytic Diels-Alder Reaction with Simple α,β-Unsaturated Ketones in: J. Am. Chem. Soc. 2002, 124, 2458–2460, doi:10.1021/ja017641u.
Literatur
- M. S. Taylor, E. N. Jacobsen: Asymmetric Catalysis by Chiral Hydrogen-Bond Donors, in: Angew. Chem. Int. Ed. 2006, 45, 1520–1543; doi:10.1002/anie.200503132.
- S. J. Connon: Organocatalysis Mediated by (Thio)urea Derivatives, in: Chem. Eur. J. 2006, 12, 5418 –5427; doi:10.1002/chem.200501076.
- B. List (Hrsg.): Organocatalysis, Thematische Serie (Open Access) im Beilstein Journal of Organic Chemistry.
Weblinks
- Informationsquellen, Daten, Beschreibung, Hintergrundinfos zum Thema Organokatalyse
- A. Berkessel, H. Gröger: Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim 2005. ISBN 978-3-527-30517-9