Gluconeogenese
Die Gluconeogenese (latinisierte Schreibung der Glukoneogenese, eines Kompositums aus altgriechisch γλυκύς glykys „süß“, νέος neos „neu“ und γένεσις genesis „Erzeugung“) ist die Bildung von D-Glucose aus organischen Nicht-Kohlenhydratvorstufen wie Pyruvat, Oxalacetat und Dihydroxyacetonphosphat. Der Stoffwechselweg ist universell bei allen Lebewesen anzutreffen. Die Ausgangsstoffe sind beim Menschen und bei Wirbeltieren Aminosäuren, die aus dem Abbau von Proteinen stammen. Pflanzen, Pilze, die meisten Bakterien und manche Invertebraten können durch den Glyoxylatzyklus Glucose auch aus Acetyl-CoA und damit aus Fettsäuren herstellen.
| Übergeordnet |
| Stoffwechsel der Glucose |
| Gene Ontology |
|---|
| QuickGO |
Notwendigkeit der Gluconeogenese beim Menschen
Der tägliche Glucosebedarf eines erwachsenen Menschen beträgt im Ruhezustand ungefähr 200 g, wobei davon allein 75 % vom Gehirn, ein Großteil des Restes von Erythrozyten genutzt werden. Die Menge an Glykogen, die im Körper gespeichert ist, beträgt etwa 400 bis 450 g. Davon sind etwa zwei Drittel in der Muskulatur gespeichert und etwa ein Drittel in der Leber. Die verfügbare Menge an Glucose im Blut beträgt etwa 5 mMol/L, was etwa 900 mg/L also 90 mg/dL entspricht.
Die Erythrozyten des Menschen und der Säugetiere besitzen keine Mitochondrien und sind daher zur Energiegewinnung vollständig auf die Zufuhr von Glucose angewiesen, welche sie über die Glykolyse und anschließende Milchsäuregärung abbauen. Das Gehirn deckt seinen enormen Bedarf an schnell verfügbarer Energie hauptsächlich ebenfalls durch Glucose-Abbau. Vor allem deshalb setzt bereits bei relativ kurzzeitigen Hungerperioden die Synthese von Glucose ein, welche vor allem in der Leber und in der Nierenrinde stattfindet und weniger im Gehirn, Skelett- und Herzmuskel. Durch den Aufbau von Glucose in der Gluconeogenese sinkt die Glucosekonzentration nie unter 3,5 mMol/L (etwa 600 mg/L, 60 mg/dL). Pro Tag können etwa 180 bis 200 g Glucose gebildet werden.
Ablauf der Gluconeogenese
Zelluläre Lokalisation
Der Ablauf der Gluconeogenese ist bei Eukaryoten auf drei Kompartimente einer Zelle verteilt. Der überwiegende Teil findet im Cytosol statt. Ein Reaktionsschritt erfolgt im Mitochondrium, ein weiterer im glatten Endoplasmatischen Retikulum (sER, nach englisch smooth endoplasmic reticulum), da das dafür jeweils notwendige Enzym (Pyruvat-Carboxylase bzw. Glucose-6-Phosphatase) nur hier vorhanden ist.
Reaktionsschritte
Ausgangsstoffe der Gluconeogenese sind entweder (1) Pyruvat oder Oxalacetat als Produkte des Aminosäureabbaus und der Milchsäuregärung (aus Lactat), (2) Pyruvat anaerob im Muskel gebildet (Cori-Zyklus), (3) Dihydroxyacetonphosphat als Derivat des Glycerins aus dem Fettabbau oder (4) Propionat, welches beim Abbau ungeradzahliger Fettsäuren nach dem letzten Schritt der β-Oxidation zurück bleibt. Dieses wird von der Propionyl-CoA-Carboxylase und einer Racemase (der Methylmalonyl-CoA-Epimerase) zu Succinyl-CoA umgesetzt, aus dem im Zuge des Citratzyklus Oxalacetat entsteht.[1]
Im Folgenden ist der Aufbau von Glucose aus L-Lactat dargestellt:
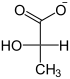 | NAD+ NADH + H+ Lactat- Dehydrogenase | 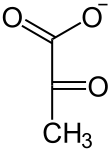 | HCO3− ATP ADP + Pi Pyruvat- Carboxylase | 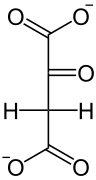 | GTP GDP +CO2 PEPCK | 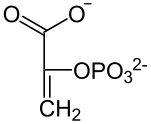 | |
| L-Lactat | Pyruvat | Oxalacetat | Phosphoenolpyruvat |
| +H2O Enolase | 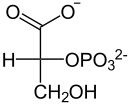 | Phospho- glycerat- Mutase | 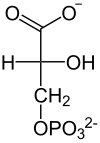 | ATP ADP Phospho- glycerat- kinase | 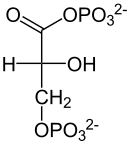 |
| Phosphoenolpyruvat | D-2-Phosphoglycerat | D-3-Phosphoglycerat | D-1,3-Bisphosphoglycerat |
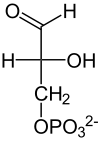
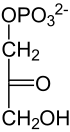
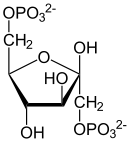 | H2O Pi Fructose-1,6-bisphosphatase | 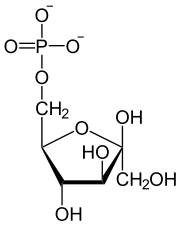 | Glucose-6-phosphat-Isomerase | 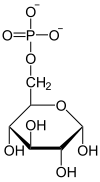 | H2O Pi Glucose-6-Phosphatase | 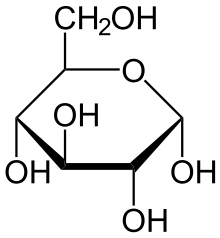 |
| β-D-Fructose-1,6-bisphosphat | β-D-Fructose-6-phosphat | α-D-Glucose-6-phosphat | α-D-Glucose |
Die Gluconeogenese entspricht nur teilweise der Umkehrreaktion der Glykolyse. Bei der Glykolyse gibt es aber drei Reaktionen, bei denen das chemische Gleichgewicht fast ausschließlich auf der Seite der Reaktionsprodukte liegt. Diese Schritte, alle von Kinasen katalysiert, sind:
- die Umwandlung von Glucose in Glucose-6-phosphat,
- von Fructose-6-phosphat in Fructose-1,6-bisphosphat und
- die Reaktion von Phosphoenolpyruvat (PEP) zu Pyruvat.
Um diese Reaktionen umzukehren, müsste die Zelle in der Lage sein, extreme Konzentrationsverhältnisse aufzubauen. Daher sind diese drei Schritte in der Glykolyse de facto irreversibel und werden in der Gluconeogenese in umgekehrter Reihenfolge wie folgt umgangen:
- die Carboxylierung von Pyruvat zu Oxalacetat unter ATP-Verbrauch (Pyruvat-Carboxylase) und die anschließende phosphorylierende Decarboxylierung von Oxalacetat zu PEP unter GTP-Verbrauch (Phosphoenolpyruvat-Carboxykinase);
- die Fructose-1,6-bisphosphatase katalysiert die Reaktion von Fructose-1,6-bisphosphat zu Fructose-6-phosphat;
- Glucose-6-phosphat wird von der Glucose-6-Phosphatase zu Glucose umgesetzt (in der Glykolyse katalysiert eine Hexokinase bzw. die Glucokinase (Hexokinase IV) die Rückreaktion).
Die anderen Umwandlungsprozesse befinden sich im Gleichgewicht, weshalb diese auch bei der Gluconeogenese eine Rolle spielen.
Ein weiterer wichtiger Unterschied zur Glykolyse ist der Reaktionsort. Während diese ausschließlich im Cytosol abläuft, ist die Gluconeogenese auf drei Kompartimente verteilt. Die Umwandlung von Pyruvat in Oxalacetat erfolgt im Lumen des Mitochondriums. Oxalacetat kann die innere Membran des Mitochondriums aber nicht frei passieren und muss erst umgewandelt werden. Dafür stehen zwei Wege zur Verfügung. Entweder wird mitochondriales Oxalacetat in PEP durch eine mitochondriale PEP-Carboxykinase überführt. PEP verlässt dann das Mitochondrium durch ein spezielles Anionen-Shuttlesystem.[2] Im Cytoplasma wird PEP infolge der Gluconeogenese in Glucose umgesetzt.
Bei Hunger wird ein zweiter Weg für den Transport eingeschlagen. In der Leber wird L-Alanin zu Pyruvat desaminiert und dient damit als Quelle für Oxalacetat. Im Hungerzustand ist die Menge an Reduktionsmittel in Form von NADH im Cytosol niedrig und im Mitochondrium hoch.[3] Für die Gluconeogenese wird jedoch NADH im Cytosol benötigt. Um sowohl NADH wie auch Oxalacetat aus dem Mitochondrium in das Cytosol zu transportieren, wird das sogenannte Malat-Aspartat-Shuttle-System verwendet. Hierbei wird das im Mitochondrium generierte Oxalacetat durch eine mitochondriale Malatdehydrogenase zu L-Malat reduziert und kann dann durch die Innere Membran transloziert werden. Für den Transport steht neben dem Malat-Aspartat-Shuttle zusätzlich der mitochondriale Dicarboxylat-Carrier zur Verfügung. Im Cytosol oxidiert eine cytosolische Malatdehydrogenase Malat zu Oxalacetat, wobei NAD+ zu NADH reduziert wird und in der Gluconeogenese eingesetzt wird.
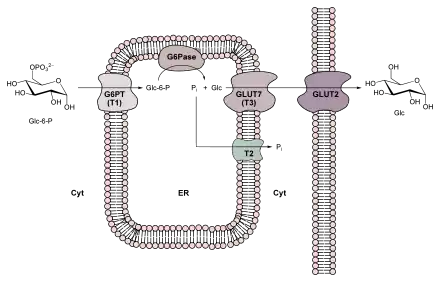
Auch der letzte Reaktionsschritt der Gluconeogenese findet nicht im Cytosol statt, sondern im Lumen des Endoplasmatischen Retikulums (ER). Den Transport ins ER und die Hydrolyse von Glucose-6-phosphat besorgt ein glucosespezifischer Membran-Enzymkomplex aus Glucose-6-phosphat-Translokase und Glucose-6-Phosphatase (vergleiche auch Abbildung rechts).
Pyruvat-Carboxylase
Die Pyruvat-Carboxylase ist nur mit ihrer prosthetischen Gruppe aktiv: Biotin. Biotin fungiert dabei als mobiler Carrier von aktiviertem Kohlenstoffdioxid. Das Biotin ist über seine Carboxygruppe an die ε-Aminogruppe eines spezifischen Lysinrestes gebunden. Dadurch entsteht ein flexibler Arm, wodurch die Biotingruppe von einem aktiven Zentrum zum zweiten „schwingen“ kann. Die Carboxylierung erfolgt in zwei Schritten:



Die erste Teilreaktion ist abhängig von der Anwesenheit von Acetyl-CoA, ohne dieses ist keine Carboxylierung von Biotin möglich. Diese Regulation ist eine Form von Allosterie, da ein hoher Acetyl-CoA-Spiegel ein Zeichen für mehr Bedarf an Oxalacetat im Citratzyklus ist. Acetyl-CoA ist ein starker und der einzige Effektor des Enzyms.[4] Oxalacetat kann entweder für die Glucogenese verwendet werden oder fließt in den Citratzyklus ein. Damit ist die katalysierte Reaktion der Pyruvat-Carboxylase ein Beispiel einer anaplerotischen Reaktion. Bei ATP-Überschuss wird das Oxalacetat in der Gluconeogenese verbraucht, wodurch dieses nicht angereichert wird. Der zweite Reaktionsschritt der Pyruvat-Carboxylase ist Acetyl-CoA unabhängig.
Vergleich Gluconeogenese und Glykolyse
Energiebilanz im Vergleich zur Umkehrung der Glykolyse
Für die Biosynthese von einem Molekül Glucose werden ausgehend vom Pyruvat vier Moleküle ATP und je zwei Moleküle GTP und NADH benötigt.


Durch die unten aufgeführte Bilanz wird deutlich, dass die obere Reaktion bevorzugt ablaufen wird, da eine direkte Umkehrung der Glykolyse eine thermodynamisch ungünstige Reaktion darstellt:

Damit sind sechs ATP-Äquivalente (2 GTP + 4 ATP) nötig, damit die Gluconeogenese zum Aufbau von einem Molekül Glucose ablaufen kann.
Gluconeogenese und Glykolyse – reziproke Regulation
Die Gluconeogenese und die Glykolyse teilen sich mehrere enzymatische Reaktionen, sind aber zwei völlig entgegenlaufende Stoffwechselwege. Daher besteht die Notwendigkeit einer Regulation. Sie findet an zwei Stellen statt:
- bei den Reaktionen vom Pyruvat zum PEP und
- bei der Umsetzung von Fructose-1,6-bisphosphat zu Fructose-6-phosphat.
Zur ersten Reaktion: die in der Glykolyse vorkommende Umwandlung von PEP in Pyruvat wird von der Pyruvatkinase katalysiert. Die Aktivität dieses Enzyms wird durch Fructose-1,6-bisphosphat erhöht und durch ATP und Alanin inhibiert. Die Enzyme der Gluconeogenese (Pyruvatcarboxylase und PEP-Carboxykinase) werden durch Acetyl-CoA aktiviert und durch ADP gehemmt. Da ATP durch Hydrolyse in ADP umgewandelt wird, kann man bei dieser Art der Regulation zweier gegenläufiger Reaktionen von reziproker Regulation sprechen. Ein weiteres Beispiel bietet hierfür die unter 2. aufgeführte Reaktion. Die bei der Glykolyse beteiligte Phosphofructokinase wird durch Fructose-2,6-bisphosphat und Adenosinmonophosphat (AMP) stimuliert, jedoch unter anderem durch Citrat inhibiert. Reziprok dazu findet die Regulation der an der Gluconeogenese beteiligten Fructose-1,6-bisphosphatase statt (durch Citrat aktiviert und durch Fructose-2,6-bisphosphat und AMP gehemmt).
Literatur
- Geoffrey Zubay: Biochemie. 4. Auflage. Mcgraw-Hill Professional, 1999, ISBN 3-89028-701-8.
- Donald Voet, Judith G. Voet: Biochemie. Wiley-VCH, 1994, ISBN 3-527-29249-7.
- Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemie. 6. Auflage. Spektrum Akademischer Verlag, Heidelberg 2007, ISBN 978-3-8274-1800-5.
- H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn, Carsten Biele (Übersetzer): Biochemie. 4., aktualisierte Auflage. Pearson Studium, 2008, ISBN 978-3-8273-7312-0.
- Reginald Garrett, Charles M. Grisham: Biochemistry. (International Student Edition). 4. Auflage. Cengage Learning Services, 2009, ISBN 978-0-495-11464-2.
- David L. Nelson, Michael M. Cox, Albert L. Lehninger (Begr.): Lehninger Biochemie. 4., vollst. überarb. u. erw. Auflage. Springer, Berlin 2009, ISBN 978-3-540-68637-8.
Weblinks
Einzelnachweise
- Gerd P. Püschel, Hartmut Kühn, Thomas Kietzmann, Wolfgang Höhne, Bruno Christ: Taschenlehrbuch Biochemie. 1. Auflage. Georg Thieme Verlag, 2018, ISBN 9783132429031, S. 252.
- B. H. Robinson: Transport of phosphoenolpyruvate by the tricarboxylate transporting system in mammalian mitochondria. In: FEBS Lett. 14 (5), 1971, S. 309–312. PMID 11945784.
- S. Jitrapakdee, M. St. Maurice u. a.: Structure, mechanism and regulation of pyruvate carboxylase. In: Biochem J. 413 (3), 2008, S. 369–387. PMID 18613815; doi:10.1042/BJ20080709.
- H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn, Carsten Biele (Übersetzer): Biochemie. 4., aktualisierte Auflage. Pearson Studium, 2008, ISBN 978-3-8273-7312-0, S. 483.
- Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemie. 6 Auflage. Spektrum Akademischer Verlag, Heidelberg 2007, ISBN 978-3-8274-1800-5, S. 518.