β-Oxidation
Als β-Oxidation bezeichnet man den biochemischen Abbaumechanismus der Fettsäuren. Die Bezeichnung bezieht sich auf die am β-C-Atom der Fettsäure stattfindenden Oxidationen. Die β-Oxidation wurde früher auch als Fettsäurespirale bezeichnet.
| Übergeordnet |
| Fettsäureoxidation |
| Untergeordnet |
| via Acyl-CoA-Dehydrogenase via Acyl-CoA-Oxidase |
| Gene Ontology |
|---|
| QuickGO |
Die β-Oxidation wurde schon 1904 von Franz Knoop in Freiburg entdeckt. Erst 50 Jahre später indes wurde der genaue Mechanismus dieses Stoffwechselweges aufgeklärt. Die β-Oxidation erfolgt bei tierischen Zellen größtenteils in den Mitochondrien, bei pflanzlichen Zellen in den Glyoxysomen.
An der β-Oxidation sind Proteine der folgenden Gene beteiligt: CPT1A, CPT1B, CPT1C, CPT2, HSD17B4, ECH1, HADHA, HADHB, ECHS1, EHHADH, ECI1, HADH, CROT.[1]
Vorbereitung
Bevor die eigentliche β-Oxidation beginnen kann, müssen die sonst sehr reaktionsträgen Fettsäuren zunächst im Zytosol "aktiviert" und anschließend vom Zytosol in die Matrix der Mitochondrien transportiert werden, wo die β-Oxidation stattfindet.
Aktivierung der Fettsäure
Ziel der Aktivierung ist die Bildung von Acyl-CoA durch Übertragung der Fettsäure auf Coenzym A. Hierbei entsteht eine energiereiche Thioesterbindung, die die weiteren Reaktionsschritte ermöglicht. Im ersten Schritt wird dazu ATP zu Pyrophosphat und AMP gespalten, das direkt zur Bildung von Acyl-AMP (auch: Acyl-Adenylat) genutzt wird. Parallel zur Spaltung des Pyrophosphats in einfaches Phosphat durch eine Pyrophosphatase kann die Fettsäure unter Abspaltung des AMP durch die frei werdende Energie mit Coenzym A verestert werden. Die so aktivierte Form der Fettsäure nennt man Acyl-CoA. Beide Reaktionen werden von einer Fettsäure-CoA-Ligase katalysiert.
Transport in die mitochondriale Matrix
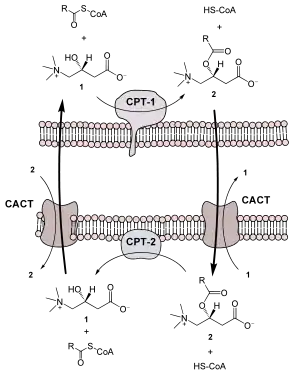
Danach wird die Acylgruppe unter Abspaltung des Coenzym A durch das Enzym Carnitin-Acyltransferase I auf Carnitin übertragen und aktiv in die Matrix der Mitochondrien transportiert. Dieser Vorgang wird durch den Carnitin-Acylcarnitin-Transporter (CACT) katalysiert, der im Antiport Acyl-Carnitin in die mitochondriale Matrix hinein und gleichzeitig Carnitin heraus befördert. In der Matrix wird der Acylrest durch die Carnitin-Acyltransferase II von Carnitin abgelöst und zurück auf Coenzym A übertragen. Während die aktivierte Fettsäure nun dem Abbau zur Verfügung steht wird das Carnitin durch den CACT wieder ins Zytosol exportiert. Die Acyl-CoA-Aktivierung ist nicht reversibel: eine aktivierte Fettsäure wird abgebaut.
Eigentliche β-Oxidation
Je nach Art der Fettsäure (Anzahl der C-Atome, Lage und Konfiguration etwaiger Doppelbindungen) kann sich der Ablauf des Abbaus von dem der geradzahligen, gesättigten Fettsäuren unterscheiden, da gegebenenfalls zusätzliche Reaktionen notwendig sind, um geeignete Substrate für die Enzyme der β-Oxidation zu schaffen oder weil andere Reaktionsprodukte als Acetyl-CoA anfallen.
Abbau geradzahliger, gesättigter Fettsäuren
Der eigentliche Abbau kann in vier aufeinander folgende Schritte unterteilt werden:
FAD-abhängige Oxidation
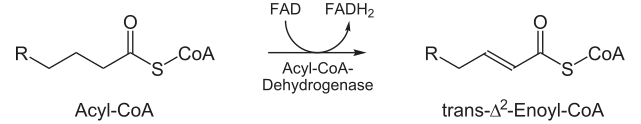
- Am Acyl-CoA wird durch das Enzym Acyl-CoA-Dehydrogenase zwischen Kohlenstoffatom 2 (Cα) und 3 (Cβ) eine trans-Doppelbindung geknüpft. Dies ist für ungesättigte Fettsäuren ungewöhnlich, die sonst meist in cis-Konfiguration vorliegen, jedoch notwendig, da das Enzym des nächsten Schrittes, die Enoyl-CoA-Hydratase, nur Fettsäuren in trans-Konfiguration erkennt. Bei diesem Vorgang wird außerdem ein FAD zu FADH2 reduziert.
Hydratisierung
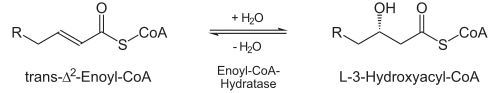
- Durch die Enoyl-CoA-Hydratase wird stereospezifisch Wasser an die neu entstandene Doppelbindung addiert, und zwar an das β-C-Atom. Es entsteht hierdurch L-3-Hydroxyacyl-CoA (auch: L-β-Hydroxyacyl-CoA).
NAD+-abhängige Oxidation
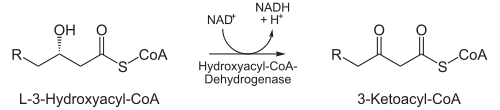
- In der nächsten Reaktion wird die C3-Hydroxygruppe durch L-3-Hydroxyacyl-CoA-Dehydrogenase (auch: β-Hydroxyacyl-CoA-Dehydrogenase) zu einer Ketogruppe oxidiert. Cofaktor hierbei ist NAD+, das die entstehenden Elektronen aufnimmt und so zu NADH + H+ reduziert wird. Dieser Schritt ist der namensgebende für den gesamten Mechanismus. Ein Beispiel ist die Dehydrierung von 2-Hydroxystearat zu 2-Oxostearat durch die 2-Hydroxyfettsäure-Dehydrogenase.
Thiolyse
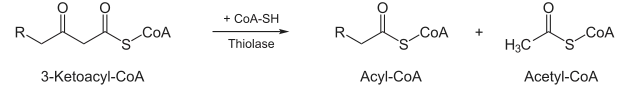
- Unter Aufnahme eines Coenzym A spaltet das Enzym 3-Keto-Thiolase Acetyl-CoA (aktivierte Essigsäure) ab, und ein um zwei Kohlenstoffatome verkürztes Fettsäuremolekül (in Form von Acyl-CoA) bleibt zurück, das wieder dem ersten Schritt zugeführt werden kann.
- Diese Reaktionsabfolge wiederholt sich so lange, bis zum Schluss zwei Acetyl-CoA übrigbleiben.
Abbau ungeradzahliger Fettsäuren
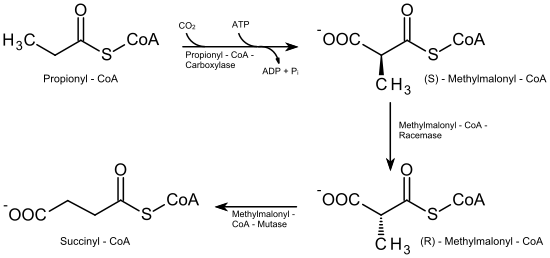
Der Abbau dieser Fettsäuren unterscheidet sich von dem der geradzahligen dadurch, dass zum Schluss nicht Acetyl-CoA, sondern Propionyl-CoA übrig bleibt. Dieses wird nun in mehreren Schritten zu Succinyl-CoA, einem Metaboliten des Citrat-Zyklus, umgebaut.
Dazu wird das Propionyl-CoA zunächst unter Spaltung eines ATP am α-C-Atom carboxyliert. Katalysiert wird diese Reaktion durch die Propionyl-CoA-Carboxylase, die als Cofaktor Biotin (Vitamin B7) enthält. Es entsteht (S)-Methylmalonyl-CoA, das im nächsten Schritt durch die Methylmalonyl-CoA-Racemase in (R)-Methylmalonyl-CoA umgewandelt wird. Zuletzt wird die Carboxygruppe durch die Methylmalonyl-CoA-Mutase, Vitamin-B12-abhängig, vom α-C-Atom auf das Kohlenstoffatom der Methylgruppe übertragen, wodurch Succinyl-CoA gebildet wird, das dem Citrat-Zyklus zugeführt werden kann.
Abbau ungesättigter Fettsäuren
Da die meisten Doppelbindungen der natürlich vorkommenden ungesättigten Fettsäuren eine cis-Konfiguration aufweisen, die Enzyme der β-Oxidation aber nur Substrate in trans-Konfiguration akzeptieren, müssen diese zunächst durch spezifische Isomerasen umgewandelt werden. Ein weiteres Problem stellen direkt aufeinander folgende Doppelbindungen (-CH=CH-CH=CH-) dar. Diese müssen so reduziert werden, dass nur noch eine Doppelbindung (-CH2-CH=CH-CH2-) bestehen bleibt, um von den Enzymen erkannt zu werden.
Energieausbeute
Das bei der β-Oxidation gebildete Acetyl-CoA kann entweder im Citrat-Zyklus weiter abgebaut, oder zur Synthese von Ketokörpern genutzt werden. Im Falle des Abbaus entstehen pro Runde der β-Oxidation ein FADH2 und ein NADH + H+, die über die Atmungskette 1,5 bzw. 2,5 ATP liefern. Jedes Acetyl-CoA, das über den Citrat-Zyklus abgebaut wird, ermöglicht zusätzlich die Synthese von 10 ATP. So können beim vollständigen Abbau eines Moleküls Palmitinsäure beispielsweise 106 Moleküle ATP gebildet werden: Palmitinsäure enthält 16 Kohlenstoffatome und wird daher zu insgesamt acht Acetyl-CoA abgebaut, wobei je sieben Moleküle FADH2 und NADH + H+ gebildet werden, da der Zyklus sieben Mal durchlaufen wird. Da zur Aktivierung der Fettsäure im Cytosol jedoch ein ATP unter Hydrolyse von zwei energiereichen Verbindungen zu AMP gespalten wurde, ergibt sich netto: 7 × 4 + 8 × 10 - 2 = 106 ATP. Im Vergleich dazu entstehen beim vollständigen Abbau von einem Molekül Glucose nur 32 Moleküle ATP.
β-Oxidation in anderen Organellen
Fettsäuren werden nicht nur in den Mitochondrien abgebaut. Bei Pflanzen und Hefen erfolgt beispielsweise der Abbau von Fettsäuren ausschließlich in den Glyoxysomen bzw. Peroxisomen.[2] Beim Menschen werden sehr langkettige Fettsäuren (mindestens 22-C-Atome) zunächst in den Peroxisomen zu kürzerkettigen Produkten abgebaut. Auch längerkettige, seltene Fettsäuren (26 bis 28 Kohlenstoffatome mit mehreren Doppelbindungen) werden durch Peroxisomen von Gehirnzellen metabolisiert.[3] Diese verkürzten Fettsäuren können dann durch die mitochondriale β-Oxidation wie oben beschrieben metabolisiert werden.
Für den Transport langkettiger Fettsäuren in das Peroxisom des Menschen wird statt Carnitin das ALD-Protein genutzt. Falls dieses einen Defekt trägt, führt dies zur Ausprägung einer Krankheit, der X-Adrenoleukodystrophie.[4][5]
Der Abbau der Fettsäuren in Peroxisomen hat gewisse Besonderheiten:[2] So oxidiert das erste Enzym die durch Coenzym A-aktivierte Fettsäure direkt mittels Sauerstoff. Dabei entsteht ein trans-Δ2-Enoyl-CoA und Wasserstoffperoxid (H2O2). Diese Reaktion wird von einer Acyl-CoA-Oxidase (EC 1.3.3.6) katalysiert und umgeht das Übertragen der Elektronen auf FAD (vgl. oben). H2O2 wird durch eine Katalase zu Sauerstoff und Wasser disproportioniert. Außerdem sind die Aktivitäten der beiden folgenden Enzyme (Enoyl-CoA-Hydratase; L-3-Hydroxyacyl-CoA-Dehydrogenase) in einem multifunktionalen Enzym vereinigt. Schließlich spaltet die peroxisomale Thiolase nicht Fettsäuren, deren Kettenlänge kürzer als acht C-Atome ist.
Umgekehrte β-Oxidation
Die Umkehrung der β-Oxidation findet in der Natur nicht statt, obwohl es keinen grundsätzlichen Hinderungsgrund gibt. Diese Umkehrung wäre sogar effizienter als die normale Fettsäuresynthese und könnte, in den geeigneten Mikroorganismen realisiert, Biokraftstoffe und Rohstoffe effizient produzieren. Im Modellorganismus E. coli gelang dies 2011 der Rice-Universität in Houston und stellt ein Beispiel für erfolgreiches Bioengineering dar. Dazu mussten 1. Teilwege für kürzere und längere Ketten dereguliert und zusammengesetzt werden; 2. die konkurrierende Glucosefermentation ausgeschaltet werden; 3. terminierende Enzyme für die gewünschten Produkte (Acyl-CoA-Reductase, Aldehyd-/Alkoholdehydrogenase, Thioesterase) eingefügt/überexprimiert werden und 4. initiierende Enzyme (Thiolasen) für die gewünschten Edukte hinzugefügt werden.[6]
Einzelnachweise
- uniprot.org
- Donald Voet, Judith G. Voet: Biochemistry. 3. Auflage. Wiley & Sons, 2004, ISBN 0-471-19350-X, S. 927.
- Geoffrey Zubay: Biochemie. 4. Auflage. Mcgraw-Hill Professional, 1999, ISBN 3-89028-701-8, S. 488.
- S. Kemp, R. J. Wanders: X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism, ABC half-transporters and the complicated route to treatment. In: Mol Genet Metab. 90 (3) 2007, S. 268–276. PMID 17092750, doi:10.1016/j.ymgme.2006.10.001
- H. W. Moser u. a.: X-linked adrenoleukodystrophy. In: Nat Clin Pract Neurol. 3 (3) 2007, S. 140–151. PMID 17342190, doi:10.1038/ncpneuro0421
- C. Dellomonaco, J. M. Clomburg u. a.: Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. In: Nature. Band 476, Nummer 7360, August 2011, S. 355–359. doi:10.1038/nature10333. PMID 21832992.
Literatur
- Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Biochemie. 6. Auflage. Spektrum Akademischer Verlag, Heidelberg 2007, ISBN 978-3-8274-1800-5.
- H. Robert Horton, Laurence A. Moran, K. Gray Scrimgeour, Marc D. Perry, J. David Rawn, Carsten Biele (Übersetzer): Biochemie. 4., aktualisierte Auflage. Pearson Studium, 2008, ISBN 978-3-8273-7312-0, S. 667ff.
- Joachim Rassow, Karin Hause, Roland Netzker, Rainer Deutzmann: Duale Reihe – Biochemie. 1. Auflage. Thieme, 2006, ISBN 3-13-125351-7.
Weblinks
- Pedro Silva: The chemical logic behind fatty acid metabolism. (engl.)