Pyruvatkinase
Die Pyruvatkinase (Pk) ist ein Enzym. Sie überträgt eine Phosphatgruppe von Phosphoenolpyruvat unter Bildung von Pyruvat auf ADP und stellt so die bei der Glykolyse gewonnene Energie als ATP bereit. Die Pk ist neben der Hexokinase und der Phosphofructokinase eines der drei regulierbaren Enzyme in der Glykolyse.[1] Sie ist daher unentbehrlich im Stoffwechsel aller Lebewesen. Während Bakterien zwei Isoformen des Enzyms besitzen, sind es bei den Wirbeltieren vier, wobei je zwei von einem Gen kodiert werden. Beim Menschen heißen diese Gene PKLR und PKM2. Mutationen im PKLR-Gen können hämolytische Anämie verursachen.[2]
| Pyruvatkinase | ||
|---|---|---|

| ||
| Pyruvatkinase M2 tetramer, Human PDB 1T5A. | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 543/574/530/530 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Homotetramer | |
| Kofaktor | Magnesium, Kalium | |
| Isoformen | L, R, M1, M2 | |
| Bezeichner | ||
| Gen-Namen | PKLR ; PKM2 | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 2.7.1.40, Kinase | |
| Reaktionsart | Übertragung einer Phosphatgruppe | |
| Substrat | ADP + Phosphoenolpyruvat | |
| Produkte | ATP + Pyruvat | |
Die vier Isoformen sind in verschiedenen Gewebetypen lokalisiert: L als das Haupt-Isozym in der Leber; R in Erythrozyten; M1 in Muskeln, Herz und Gehirn; und M2 im Fötus.[2]
Reaktion
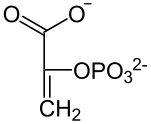 + ADP
+ ADP
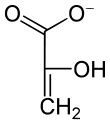 + ATP
+ ATP
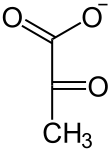 + ATP
+ ATP


Durch die Katalyse wird vom Phosphoenolpyruvat (PEP) eine Phosphatgruppe auf ADP übertragen. Es entstehen ATP und Pyruvat. Letzteres wird über den Pyruvat-Dehydrogenase-Komplex oxidativ decarboxyliert zu Acetyl-CoA, welches im Citratzyklus weiter abgebaut wird. Dabei entsteht jedoch nicht Pyruvat direkt, sondern das im Gleichgewicht stehende Enolpyruvat.[3] Bei pH 7 liegt das Gleichgewicht auf Seiten der Ketoform. Die Keto-Enol-Tautomerie verläuft spontan
( = −35 bis −40 kJ/mol) und ist zum größten Teil für die Änderung der freien Energie durch die Hydrolyse von PEP verantwortlich.[4]

Die Gluconeogenese kann aus thermodynamischen Gründen nicht über die Pyruvatkinase (wie bei der Glykolyse) verlaufen und daher werden die Enzyme Pyruvatcarboxylase, die unter anderem Oxalacetat für den ersten Schritt des Citratzyklus bereitstellt (anaplerotische Reaktion), und Phosphoenolpyruvat-Carboxykinase verwendet.[1]
Struktur
Die Pyruvatkinase des Menschen ist ein Tetramer, bestehend aus vier identischen Protein-Untereinheiten mit je 528 Aminosäuren.[5]
In Säugetieren gibt es vier Isoformen des Enzyms:
- L in Leber und Nieren
- R in Erythrozyten
- M1 in Muskeln, Herz, Gehirn, Leukozyten und Thrombozyten
- M2 in frühem fetalen Gewebe und in Tumorgewebe
L und R sowie M1 und M2 stammen jeweils von demselben Gen, werden jedoch durch unterschiedliche Promotoren transkribiert.[6]
Regulation der Pyruvatkinase
Beide Formen der Pyruvatkinase werden durch Fructose-1,6-bisphosphat aktiviert. Fructose-1,6-bisphosphat entsteht bei der dritten Reaktion der Glykolyse, welche durch Phosphofructokinase katalysiert wird. Diese Reaktion, der sog. committed step, bestimmt durch ihr Reaktionsprodukt, das Fructose-1,6-bisphosphat, welches als Aktivator für die nächste und letzte Reaktion der Glykolyse agiert, die Reaktionsgeschwindigkeit der Glykolyse. Diese Regulation ermöglicht eine Homöostase der Intermediärprodukte der Glykolyse.
Durch eine hohe Energieladung in der Zelle (hohe ATP-Konzentration) und die Anwesenheit von Alanin wird die Pyruvatkinase inhibiert. Somit läuft die Reaktion nicht ab, wenn keine weitere Energie von der Zelle benötigt wird.
Das L-Isozym wird zusätzlich durch Proteinphosphorylierung kontrolliert. Ist der Glucosespiegel im Blut niedrig, bewirkt das Hormon Glucagon die Phosphorylierung der Pyruvatkinase, welche dadurch an Aktivität verliert. So bleibt Phosphoenolpyruvat erhalten und steht für die Gluconeogenese zur Verfügung, durch welche neue Glucose in der Leber aufgebaut wird. Auch intrazelluläres Calcium lässt Pyruvatkinase phosphorylieren.
Pyruvatkinase-Defekte
Das PKLR-Gen liegt auf dem Genlocus 1q22 (Chromosom 1).[7] Nur vom PKLR-Allel der Pyruvatkinase sind Mutationen bekannt und nur bei der R-Form zeigen sich diese als Defekt. Mangel an PKR (Pyruvatkinasemangel PK) ist eine autosomal-rezessiv vererbte Stoffwechselstörung. Dadurch wird häufig eine hämolytische Anämie verursacht, da die roten Blutkörperchen nicht genügend ATP für ihre Membranpumpen erzeugen können.[8] Überschuss an PKR-Aktivität mit entsprechend erhöhtem ATP in Blutzellen ist als weiterer Defekt bekannt.[2]
Einzelnachweise
- Abbau und Synthese der Glucose – Wissen für Mediziner. Abgerufen am 6. März 2020.
- UniProt P30613
- David Nelson, Michael Cox: Lehninger Biochemie. Springer, Berlin; 4., vollst. überarb. u. erw. Auflage. 2008, ISBN 978-3-540-68637-8, S. 713.
- Reginald H. Garrett, Charles M. Grisham: Biochemistry. Cengage Learning, 2008, ISBN 978-0-495-10935-8, S. 551 (eingeschränkte Vorschau in der Google-Buchsuche).
- G. Valentini, L. R. Chiarelli u. a.: Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. In: The Journal of biological chemistry. Band 277, Nummer 26, Juni 2002, S. 23807–23814, ISSN 0021-9258. doi:10.1074/jbc.M202107200. PMID 11960989.
- Eintrag zu Pyruvat-Kinase. In: Römpp Online. Georg Thieme Verlag, abgerufen am 13. September 2013.
- PKLR in der humanen Datenbank HGNC
- Todd A. Swanson, Sandra I. Kim und Marc J. Glucksman: BRS Biochemistry, Molecular Biology, and Genetics. Lippincott Raven; 5. Auflage 2010; ISBN 978-0-7817-9875-4; S. 68