Aldehyd-Dehydrogenase 2
Aldehyddehydrogenase 2 (ALDH-2) ist ein zur Gruppe der Aldehyddehydrogenasen gehörendes Enzym, welches im menschlichen Körper zum Abbau von Alkohol (Ethanol) benötigt wird. ALDH-2 wandelt den – durch ADH aus Alkohol erzeugten – toxischen Acetaldehyd (Ethanal) in Acetat (Ethansäure) um.

Mechanismus der Umwandlung von Aldehyden in Carbonsäuren durch die Aldehyddehydrogenase
| Aldehyd-Dehydrogenase 2 | ||
|---|---|---|
| ||
| Oktamer nach PDB 1CW3 | ||
|
Vorhandene Strukturdaten: 1a4z, 1ag8, 1cw3, 1nzw, 1nzx, 1nzz, 1o00, 1o01, 1o02, 1o04, 1o05, 1of7, 1zum, 2onm, 2onn, 2ono, 2onp | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 500 Aminosäuren; 54,4 kDa | |
| Sekundär- bis Quartärstruktur | Homotetramer | |
| Isoformen | ALDH1 | |
| Bezeichner | ||
| Gen-Namen | ALDH2 ; ALDH-E2; ALDHI; ALDM; MGC1806 | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 1.2.1.3, Oxidoreduktasen | |
| Substrat | Aldehyd + NAD+ + H2O | |
| Produkte | Carbonsäure + NADH | |
| Vorkommen | ||
| Homologie-Familie | HBG439831 | |
| Übergeordnetes Taxon | Lebewesen | |
| Orthologe | ||
| Mensch | Maus | |
| Entrez | 217 | 11669 |
| Ensembl | ENSG00000111275 | ENSMUSG00000029455 |
| UniProt | P05091 | Q3TVM2 |
| Refseq (mRNA) | NM_000690 | NM_009656 |
| Refseq (Protein) | NP_000681 | NP_033786 |
| Genlocus | Chr 12: 110.69 – 110.73 Mb | Chr 5: 121.83 – 121.85 Mb |
| PubMed-Suche | 217 | 11669 |
Eigenschaften
Die Aldehyd-Dehydrogenase ist hauptsächlich als ein alkoholabbauendes Enzym bekannt. Seine mitochondriale Isoform, die mtALDH2, wirkt in erster Linie schützend auf den Herzmuskel. Seine kardioprotektive Aktivität ist nach einer Sauerstoffmangelsituation im Herzen, z. B. durch einen Verschluss der Koronargefäße, für das Überleben der Herzmuskelzellen von hoher Bedeutung.[1] Die ADH2 schützt das Gewebe und die Zellen vor der schädigenden Wirkung von Alkohol bzw. Acetaldehyd, und anderen toxischen Aldehyden. mtALDH2 spielt eine wichtige Rolle in der Detoxifikation der reaktiven Sauerstoffspezies (engl.: Reactive Oxygen Species, ROS) und ist dadurch maßgeblich an der Aufrechterhaltung einer intakten Funktion der Mitochondrien beteiligt.[2] Es kommt daher sehr häufig im Herz vor, einem Organ das besonders viele Mitochondrien enthält und deshalb empfindlich gegenüber oxdidativem Stress und ROS ist.[3] Die Aktivierung von mtALDH2 beugt hauptsächlich der vorzeitigen Apoptose und Nekrose der Herzmuskelzellen[2] sowie der Bildung von fibrotischen Gewebes vor.[3]
46 Prozent der Japaner und 56 Prozent der Chinesen sind von einem Polymorphismus der Acetaldehyddehydrogenase 2 betroffen. Sie sind Träger eines dominanten Allels des ALDH2-Gens, bei dem an Position 487 der Aminosäuresequenz das Glutamat gegen Lysin ausgetauscht ist. Das veränderte ALDH2 kann Acetaldehyd weniger effektiv verarbeiten und wird selbst schneller abgebaut.[4] Dadurch kommt es leichter zu einer Anhäufung des Acetaldehyds im Körper und damit zu den mit übertriebenem Alkoholkonsum verbundenen Vergiftungserscheinungen (Flush-Syndrom). Die betroffenen Personen sind somit empfindlicher gegenüber den negativen Auswirkungen des Alkoholkonsums.
Einige Milchsäurebakterien beschreiten mit der Aldehyddehydrogenase 2 auch den entgegengesetzten Weg: Unter guten Bedingungen bauen sie das gesamte aus der Glykolyse stammende Pyruvat zu Lactat ab. Herrscht allerdings Glucosemangel, spalten verschiedene homofermentative Stämme das Pyruvat mittels Pyruvat-Formiat-Lyase außerdem in Formiat und Acetyl-Coenzym A. Die Hälfte des Acetyl-CoA kann nun von der Aldehyddehydrogenase in Acetaldehyd umgesetzt werden, das die Alkoholdehydrogenase in Ethanol umwandelt. Aus der anderen Hälfte des Acetyl-CoA wird Acetat hergestellt, das zur ATP-Synthese genutzt werden kann.
Die Rolle von ROS und die Aktivierung von mtALDH2
mtALDH2 ist ein Schlüsselenzym in der ischämischen Präkonditionierung.[1] Wird das Myokard durch kurze, für das Herz unschädliche, ischämische Phasen präkonditioniert und folgt darauf ein schwerer Infarkt, so sind die Schäden am Myokard, die durch den Sauerstoffmangel hervorgerufen werden deutlich geringer als nach einem Infarkt ohne Präkonditionierung.[5]
Ergebnis der Präkonditionierung ist die Aktivierung von mtALDH2. Eine Präkonditionierung kann auch durch chemische Substanzen wie Ethanol oder Anästhetika hervorgerufen werden. Ein wichtiger Aktivator der mtALDH2 bei der Päkonditionierung ist die Proteinkinase-ε (PKC-ε). Diese wird von den RISK (engl.: Reperfusion Injury Salvage Kinases[6]) durch Phosphorylierung aktiviert. RISK selbst werden durch verschiedene Mechanismen der Präkonditionierung aktiviert. Das durch RISK aktivierte PKC-ε wandert aus dem Zytosol in das Mitochondrium, um dort mtALDH2 durch Phosphorylierung zu aktivieren.[7]
Bildung von ROS und Lipidperoxidation
Eine besonders wichtige Aufgabe von mtALDH2 ist es, die für die Zelle schädlichen reaktive Aldehyde aus der Zelle zu entfernen.[3] Es ist an der Detoxifikation von reaktiven Aldehyden wie z. B. 4-Hydroxynonenal (4-HNE) oder Malondialdehyd (MDA) beteiligt.[2]
Reaktive Aldehyde entstehen im Myokard hauptsächlich in der Reperfusionsphase, wenn nach Auflösung einer Ischämie, das Gewebe plötzlich wieder durchblutet und mit Sauerstoff versorgt wird. Diese schlagartige Rückführung von Sauerstoff führt zu oxidativen Stress,[1] ein Zustand bei dem in der Zelle mehr ROS gebildet als abgebaut werden.[3][8] ROS oxidieren ungesättigte Fettsäuren wie z. B. Arachidonsäure und Linolsäure mit Peroxiden, wodurch u. a. die mitochondriale Membran geschädigt wird und reaktive Aldehyde wie 4-HNE entstehen. Dieser Prozess wird als Lipidperoxidation bezeichnet.[1][3] Die aus der Lipidperoxidation gebildeten Aldehyde sind durch ihre ungesättigten α/β-Kohlenstoffatome sehr reaktiv und können deshalb durch Reaktion mit den Aminosäureresten von Cystein, Histidin oder Lysin der Proteine, zellschädigende Proteinaddukte bilden.[3]
Reaktive Aldehyde hemmen die Elektronentransportkette im Mitochondrium und induzieren die Öffnung der mitochondrialen Permeabilitäts-Transitions-Pore (mPTP). Der Angriff auf die mitochondriale Membran führt außerdem zur Dysfunktion der Mitochondrien, wodurch sie noch mehr ROS produzieren. Darüber hinaus führt eine nicht intakte Mitochondrienmembran dazu, dass die an der inneren Mitochondrienmembran lokalisierten Enzyme, vor allem Cytochrom C, freigesetzt werden. Die Freisetzung von Cytochrom C führt zu Bildung eines Apoptosoms und damit zum Zelltod. mtALDH2 unterbricht die Spirale von ROS-Bildung und Mitochondrienschädigung, in dem es die Bildung von ROS sowie die Blockade der der Elektronentransportkette an Komplex I und IV sowie die Ca2+ induzierte Öffnung der mPTP hemmt[1].
Ischämie-Reperfusionsschaden- IR-Schaden: Das Geschehen, bei dem die Zellmembran und in weiterer Folge das ganze Gewebe, sowohl durch den Sauerstoffmangel während der Ischämie, als auch durch die anschließende Reperfusion des Gewebes geschädigt wird, ist die Ursache für einen Ischämie-Reperfusions-(IR)-Schaden.[5]
ROS vermittelte Aktivierung von PKC-δ
Wie schon einleitend festgestellt wurde, übt die PKC-ε über die Aktivierung der mtALDH2 eine positive Wirkung auf ein IR-Geschehen aus. Ihre Isoform, die Proteinkinase-δ (PKC-δ) beteiligt sich jedoch an gegenteiligen Vorgängen. PKC-δ wird durch ROS aktiviert und transloziert daraufhin aus dem Zytosol in das Mitochondrium.
Dort vermittelt PKC-δ über eine cAMP-abhängige Proteinkinase die aktivierende Phosphorylierung am Serin616 des Dynamin-related-Proteins-1 (Drp1). Drp1 ist für die natürliche Spaltung (Fission) zweier, im Rahmen ihrer Wiederverwertung fusionierter, Mitochondrien verantwortlich. Die gesteigerte Aktivität von Drp1 führt jedoch zu einer übermäßigen Zweiteilung und damit zu Entfernung einzeln vorliegender, zum Abbau bestimmter Mitochondrien. Neben der Phosphorylierung von Drp1, führt die Aktivierung von PKC-δ zur Aktivierung der Glykogensynthase-Kinase-3β (GSK-3β). Wird die GSK-3β eingeschaltet, so bewirkt sie u. a. die Öffnung der mPTP und damit die Induktion der Apoptose.
mtALDH2 gewinnt auch beim Vorgang der Hemmung von PKC-δ an Bedeutung, weil es unerlässlich für die Detoxifikation von ROS ist. Es beseitigt ROS und entfernt dadurch einen Aktivator von PKC-δ. Ein anderer Aspekt der Hemmung ist die Induktion von PKC-ε. Durch deren Aktivierung durch die RISK kommt es zu einer negativen Rückkopplung auf die Expression und Translokation ihres Isoenzyms PKC-δ.[8]
Kardioprotektive Wirkung von Ethanol
mtALDH2 fungiert als Enzym sowohl im Alkoholmetabolismus als auch im Abbau von reaktiven Aldehyden. Auf Grund dieser Tatsache scheint es möglich, dass sich eine Alkohol-induzierte Aktivierung von mtALDH2, positiv auf den Schutz des Herzmuskels auswirkt.
Tatsächlich lässt sich nach moderatem Alkoholkonsum eine gesteigerte Aktivität von mtALDH2 feststellen. Diese Präkonditionierung mit Alkohol führt dazu, dass bei einem IR-Geschehen eine hohe Konzentration an bereits aktivierten mtALDH2 vorliegt. Ein erhöhter Spiegel von aktiven mtALDH2 erweitert die Kapazität der Zelle, reaktive Aldehyde abzubauen und die Lipidperoxidation zu hemmen. Ein weiterer kardioprotektiver Effekt von Alkohol ist die Aktivierung der Superoxid-Dismutase (SOD). SOD ist ein wichtiges Antioxidans des Myokards, das durch Detoxifikation gefährliche ROS aus den Herzmuskelzellen beseitigt.[9] Im asiatischen Raum sind 40 % der Bevölkerung Träger einer Mutation des ALDH2 Allels. Sie besitzen den ALDH2*2-Mutanten, welcher weniger aktiv ist als der Wild-Typ. Träger von ALDH2*2 sind dadurch einem höheren Risiko ausgesetzt, kardiale Schädigungen durch chronischen Alkoholkonsum zu erleiden.[3]
Hemmung der Apoptose
Obwohl durch eine rasche Wiederversorgung mit Sauerstoff durch Reperfusion des Herzmuskels die Ausbreitung des Infarktgebietes deutlich reduziert werden kann, verursacht die Rückführung von Sauerstoff paradoxerweise selbst auch Schädigungen des Myokards (IR-Schaden).[1]
Während der Ischämischen Phase gehen die Zellen des Herzmuskels durch Nekrose zugrunde. Hingegen, während der Reperfusion sterben die Zellen durch Apoptose. Dies hängt damit zusammen, dass die Apoptose ein ATP abhängiger Prozess ist. Während der Ischämie ist die Herstellung von ATP durch Blockade der sauerstoffabhängigen ATP-Synthesewege a.v. der Hypoxie deutlich reduziert. Der Mangel an ATP verhindert damit weitgehend den Zelltod durch Apoptose. Durch die Reperfusion wird das Gewebe wieder mit Sauerstoff und Glukose versorgt. Die Rückführung von Sauerstoff sorgt einerseits dafür, dass die ATP-Konzentration wieder steigt, andererseits kommt es durch das hohe Sauerstoffangebot zu oxidativen Stress. Die Kombination aus ROS – und ATP Bildung fördert den Zelltod durch Apoptose. mtALDH2 wirkt an dieser Stelle kardioprotektiv, in dem es die Bildung von ROS hemmt und damit einer Apoptose entgegenwirkt[1]. Zusätzlich hemmt mtALDH2 einen Signalweg der Apoptose, indem es die Aktivität von Akt und von der AMP-abhängigen Proteinkinase (AMPK) steigert, welche dann ihre apoptotischen Zielenzyme Foxo3 und Caspase-3 hemmen.[3]
Autophagie
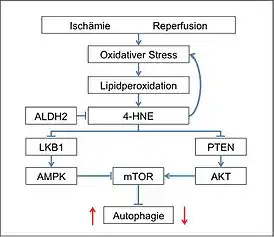
Neben Nekrose und Apoptose trägt auch die Autophagie dazu bei, dass nach einer Ischämie mit anschließender Reperfusion das Herzmuskelgewebe in seiner Funktion eingeschränkt ist. Die Autophagie ist ein natürlicher Mechanismus der Zellen bei dem durch Abbau von zelleigenen Material das Überleben und das Absterben der Zelle reguliert wird. Dieser Prozess kann jedoch von 4-HNE übermäßig gesteigert wird. 4-HNE sammelt sich durch den Einfluss von ROS in den Herzmuskelzellen an. Es reguliert die Autophagie der Herzmuskelzellen während der Ischämie und Reperfusion in entgegengesetzter Richtung.[1]
Normalerweise findet zum Schutz des Myokards während der Ischämie vermehrt Autophagie statt. Hierbei wird der Inhibitor der Autophagie, mTOR durch LKB1 (Leberkinase B1) bzw. AMPK, gehemmt, sodass die Autophagie möglich ist. Während der Reperfusion aber, wird mTOR durch PTEN bzw. Akt aktiviert, so dass mTOR nun die Autopgagie hemmt. Dadurch wird eine exzessive Autophagie während der Reperfusion verhindert. 4-HNE jedoch, blockiert sowohl PTEN als auch LKB1. Dadurch ist mTOR einerseits während der Ischämie aktiv, sodass es eine, während der Sauerstoffmangelphase vorteilhafte, Autophagie gehemmt wird. Andererseits wird es während der Reperfusion nicht aktiviert und kann deshalb die Autophagie zu diesem Zeitpunkt nicht hemmen. mtALDH wirkt der Fehlregulation von 4-HNE entgegen, in dem es die Bildung von 4-HNE eindämmt und dessen Abbau vorantreibt.[1]
Mitophagie
mtALDH2 verhindert auch, die durch oxidativen Stress gehemmte Mitophagie also den Abbau geschädigter Mitochondrien. Geschädigte Mitochondrien müssen zum Schutz der Zelle abgebaut werden. Oxidativer Stress führt zu einer Inaktivierung von Parkin, welches für den Abbau des Mitochondriums wichtig ist, und verhindert damit die Entfernung des geschädigten Mitochondriums aus der Zelle. Werden die nicht richtig funktionierenden Mitochondrien nicht abgebaut, so bilden diese mehr ROS was in weiterer Folge zu oxidativen Stress führt. mtALDH2 schützt die Zelle vor einer Inaktivierung der Mitophagie in dem es die Entstehung von ROS und die damit verbundene Inaktivierung von Parkin hemmt.[1]
ER-Stress
Durch die hypoxischen Bedingungen während der Ischämie wird der Proteinhaushalt der Myokardzellen, insbesondere die Proteinfaltung und der Proteinabbau, empfindlich gestört. Verschiedene Stressoren, wie z. B. Hypoxie führen dazu, dass die Kapazität des Endoplasmatischen Retikulums (ER), neu synthetisierte Proteine korrekt zu falten und falsch gefaltete Proteine richtig zu falten, abnimmt. Faltungsbedürftige Protein sammeln sich deshalb in der Zelle an, und es kommt zu einem Zustand der ER-Stress genannt wird.[2] ER-Stress löst im Myokard die Apoptose der Kardiomyozyten aus und reduziert das Herzmuskelgewebe somit um einen Großteil seiner funktionsfähigen Zellen.[2]
ER-Stress führt über die NADPH-Oxidase zur Apoptose. Damit die Zelle trotz ER-Stress überleben kann, wird dieser apoptotische Signalweg durch den, an einen Wachstumsfaktor gekoppelten Signalwegen von Akt und PI3K gehemmt. Hierbei wird Akt von PI3K phosphoryliert. Das nun aktivierte Akt hemmt die p47phox Untereinheit der NADPH-Oxidase. Wäre die p47phox Untereinheit nicht gehemmt, so würde sie die Apoptose einleiten. Dies geschieht, wenn durch ER-Stress die PI3K gehemmt und damit die Aktivierung von Akt verhindert wird. Somit bleibt die p47phox Untereinheit aktiv und leitet die Apoptose ein. mtALDH2 hält den Signalweg von PI3K und Akt auch während ER-Stress aufrecht. Sie hebt die hemmende Wirkung des ER-Stress auf PI3K auf, sodass Akt weiterhin aktiviert wird und die Apoptose hemmt.[2]
Die während des ER-Stresses typischerweise entstehenden Chaperone und Regulatoren wie z. B. GRP78 und CHOP vermitteln selbst auch, auf noch unbekannte Weise, die Apoptose. Die Apoptose hemmende Wirkung von mtALDH2 besteht hierbei darin, CHOP und GRP78 zu hemmen. Ein weiterer ER-Stress auslösender Stressor ist 4 HNE. mtALDH2 beseitigt diesen Stressfaktor, indem es 4-HNE durch Detoxifikation eliminiert.[2]
Fibrosebildung
Die Bildung einer Fibrose im Herz, ist die Hauptursache für eine herabgesetzte Herzleistung nach einem Infarkt. Sie geht mit einer verminderten Kontraktilität des Herzmuskels und einem verringerten Auswurfvolumen des linken Ventrikels (Left Ventricular Ejection Fraction – LVEF) einher. Ein Grund für die Veränderung des Herzmuskelgewebes ist ein überaktiver Wnt/β-Catenin-Signalweg. Dieser Signalweg ist für die Regeneration und Reparation des Gewebes verantwortlich und ist unter physiologischen Bedingungen inaktiv bzw. stark reguliert.[10] Ein Infarkt führt zu dessen andauernden und übermäßigen Aktivierung, wodurch sich das Myokard und Epikard in stark fibröses weitgehend funktionsloses Gewebe umwandelt. Die Ausbildung der kardialen Fibrose erfolgt einerseits durch die Wnt-vermittelte epithelial-mesenchymale-Transition (EMT) der Zellen des Epikards in Fibroblasten. Andererseits wird das Myokard, durch die Infiltration seiner infarktgeschädigten nekrotischen Areale mit Fibroblasten zu einem weniger elastischen und kontraktionschwächeren Gewebe umstrukturiert.[10][11]
Wnt/β-Catenin-Signalweg
In Abwesenheit von Wnt befindet sich der Transkriptions-Coaktivator β-Catenin in einem Komplex, in welchem er über Axin und dem APC- Protein (Adenomatöses Polyposis Coli) mit, der CK1 (Caseinkinase1) und GSK-3β verknüpft ist. In diesem Komplex gebunden, wird β-Catenin von CK1 und GSK-3β phosphoryliert. Das phosphorylierte β-Catenin wird von der β-TrCP Untereinheit der E3-Ubiquitin-Ligase erkannt und ubiquitiniert. Durch die Ubiquitinierung wird es im Proteasom abgebaut und somit kontinuierlich aus dem Zytosol eliminiert.
Ist Wnt anwesend, dann formiert es einen Rezeptorkomplex mit dem 7-Transmembranrezeptor Frizzled und dem Korezeptor LRP5 (Low-Density-Lipoprotein(LDL)-Receptor-related-Protein) oder LRP6. Nun binden auf der zytosolischen Seite des Rezeptorkomplexes das Protein Disheveled (Dsh), GSK-3β und CK1. Die beiden Kinasen GSK-3β und CK1 phosphorilieren LRP, wodurch Axin an LRP gebunden werden kann. Durch die Anbindung von Axin an den Rezeptorkomplex in der Membran bleibt β-Catenin unphosphoryliert im Zytosol und wird nicht abgebaut. Es wandert in den Zellkern wo es gemeinsam mit dem T-cell-factor (TCF) die Transkription seiner Zielgene einschaltet.[11] Ein bedeutendes Zielgen ist das Wnt-Inducible-Signaling Pathway (WISP)-1-Gen. WISP-1 ist ein Wachstumsfaktor der insbesondere die Synthese und Freisetzung von Kollagen sowie die Proliferation der Fibroblasten stimuliert[10].
In Anwesenheit von mtALDH2 ist die Ausbildung einer Fibrose deutlich gedrosselt. Fehlt mtALDH2, so ist die Konzentration von β-Catenin, Wnt und aktiver GSK-3β erhöht. mtALDH2 hemmt die Dephosphorylierung der GSK-3β und verhindert damit deren Aktivierung, denn phosphorylierte GSK-3β ist inaktiv. Der Prozess der Stabilisierung von β-Catenin wird gehemmt, wodurch die Signalkaskade über die Wnt seine Zielgene einschaltet, unterbrochen wird. Die Aktivierung von mtALDH2 führt außerdem dazu, dass weniger Kollagen I und III gebildet und in den Zellen angestaut wird. Zudem wird die Expression von α-SMA (engl.:Smooth Muscle Actin) und WISP-1 gehemmt. Auch sei an die Funktion von mtALDH2 als Alkoholabbauendes Enzym erinnert, denn das im Alkoholabbau entstehende und von mtALDH2 abgebaute Acetylaldehyd ist dafür bekannt, die Fibrogenese zu fördern.[10]
Aktivierung von Arzneistoffen durch ALDH2
Aldehyd-Dehydrogenase 2 ist in ihrer Eigenschaft als Nitratreduktase beteiligt an der Aktivierung zweier NO-Donatoren. Konkret katalysiert sie die Freisetzung von Stickstoffmonoxid aus Glyceroltrinitrat und Pentaerythrityltetranitrat in den Mitochondrien glatter Muskelzellen.[12] Das NO bewirkt dort eine Relaxation, wodurch eine Weitung der Gefäße eintritt.
Einzelnachweise
- Pang Jiao-Jiao, Chen You-Go, Ren Jun: Mitochondrial aldehyde dehydrogenase in myocardial ischemia-reperfusion injury: from bench to bedside. In: Acta Physiologica Sinica,. NCBI National Center for Biotechnology Information, 25. Dezember 2015, S. 335–344, abgerufen am 1. April 2017 (englisch).
- J. Liao, A. Sun, Y. Xie, T. Isse, T. Kawamoto, Y. Zou, J. Ge: Aldehyde dehydrogenase-2 deficiency aggravates cardiac dysfunction elicited by endoplasmic reticulum stress induction. In: Molecular medicine. Band 18, Juli 2012, S. 785–793, doi:10.2119/molmed.2011.00466, PMID 22430940, PMC 3409283 (freier Volltext).
- C. H. Chen, L. Sun, D. Mochly-Rosen: Mitochondrial aldehyde dehydrogenase and cardiac diseases. In: Cardiovascular research. Band 88, Nummer 1, Oktober 2010, S. 51–57, doi:10.1093/cvr/cvq192, PMID 20558439, PMC 2936126 (freier Volltext) (Review).
- Q. Xiao, H. Weiner, D. W. Crabb: The mutation in the mitochondrial aldehyde dehydrogenase (ALDH2) gene responsible for alcohol-induced flushing increases turnover of the enzyme tetramers in a dominant fashion. In: The Journal of clinical investigation. Band 98, Nummer 9, November 1996, S. 2027–2032, ISSN 0021-9738. doi:10.1172/JCI119007. PMID 8903321. PMC 507646 (freier Volltext).
- C. E. Murry, R. B. Jennings, K. A. Reimer: Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. In: Circulation. Band 74, Nr. 5, 1. November 1986, ISSN 0009-7322, S. 1124–1136, PMID 3769170.
- H. Kalakech, P. Hibert, D. Prunier-Mirebeau, S. Tamareille, F. Letournel, L. Macchi, F. Pinet, A. Furber, F. Prunier: RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. In: PLOS ONE. Band 9, Nummer 9, 2014, S. e107950, doi:10.1371/journal.pone.0107950, PMID 25237809, PMC 4169577 (freier Volltext).
- Xiao-E. Lang, Xiong Wang, Ke-Rang Zhang, Ji-Yuan Lv, Jian-Hua Jin: Isoflurane Preconditioning Confers Cardioprotection by Activation of ALDH2. In: PLOS ONE. Band 8, Nr. 2, 28. Februar 2013, ISSN 1932-6203, S. e52469, doi:10.1371/journal.pone.0052469, PMID 23468836, PMC 3585331 (freier Volltext).
- Shijun Wang, Feng Zhang, Gang Zhao, Yong Cheng, Ting Wu: Mitochondrial PKC-ε deficiency promotes I/R-mediated myocardial injury via GSK3β-dependent mitochondrial permeability transition pore opening. In: Journal of Cellular and Molecular Medicine. 1. März 2017, ISSN 1582-4934, doi:10.1111/jcmm.13121.
- Qing Yuan, Shanjuan Hong, Shu Han, Li Zeng, Fang Liu: Preconditioning with Physiological Levels of Ethanol Protect Kidney against Ischemia/Reperfusion Injury by Modulating Oxidative Stress. In: PLOS ONE. Band 6, Nr. 10, 12. Oktober 2011, ISSN 1932-6203, S. e25811, doi:10.1371/journal.pone.0025811, PMID 22022451, PMC 3192120 (freier Volltext).
- Xinjun Zhao, Yue Hua, Hongmei Chen, Haiyu Yang, Tao Zhang: Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/β-catenin signaling pathway. In: Therapeutics and Clinical Risk Management. Band 11, 11. September 2015, ISSN 1176-6336, S. 1371–1381, doi:10.2147/TCRM.S88297, PMID 26392772, PMC 4574798 (freier Volltext).
- Bryan T. MacDonald, Keiko Tamai, Xi He: Wnt/β-catenin signaling: components, mechanisms, and diseases. In: Developmental cell. Band 17, Nr. 1, 6. April 2017, ISSN 1534-5807, S. 9–26, doi:10.1016/j.devcel.2009.06.016, PMID 19619488, PMC 2861485 (freier Volltext).
- Schubert-Zsilavecz, Manfred., Roth, Hermann J.: Medizinische Chemie : Targets - Arzneistoffe - chemische Biologie ; 191 Tabellen. 2., völlig neu bearb. und erw. Auflage. Dt. Apotheker-Verl, Stuttgart 2010, ISBN 978-3-7692-5002-2.