P4-t-Bu
P4-t-Bu ist ein einfach zugänglicher Vertreter aus der Gruppe der neutralen, peralkylierten sterisch gehinderten Polyaminophosphazene, die extrem starke, aber nur sehr schwache nucleophile Basen darstellen. P4-t-Bu kann auch als tetrameres Triaminoiminophosphoran der Grundstruktur (H2N)3P=N-H aufgefasst werden. Die homologe Reihe von P1- bis P7-Polyaminophosphazenen[4][1] der allgemeinen Formel mit bevorzugt Methylgruppen als R1, einer Methylgruppe oder tert. Butylgruppe als R2 und geradzahliges x zwischen 0 und 6 (P4-t-Bu: R1 = Me, R2 = t-Bu und x = 3)[5] ist durch Arbeiten von Reinhard Schwesinger erschlossen worden; die erhaltenen Phosphazenbasen werden daher auch als Schwesinger-Superbasen bezeichnet.[6][7]

| Strukturformel | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
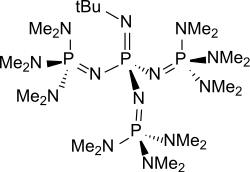 | ||||||||||||||||
| Die Methylgruppen (CH3-Gruppen) sind als Me angegeben | ||||||||||||||||
| Allgemeines | ||||||||||||||||
| Name | P4-t-Bu | |||||||||||||||
| Andere Namen |
| |||||||||||||||
| Summenformel | C22H63N13P4 | |||||||||||||||
| Kurzbeschreibung |
farblose Kristalle[1] | |||||||||||||||
| Externe Identifikatoren/Datenbanken | ||||||||||||||||
| ||||||||||||||||
| Eigenschaften | ||||||||||||||||
| Molare Masse | 633,86 g·mol−1 | |||||||||||||||
| Aggregatzustand |
fest | |||||||||||||||
| Schmelzpunkt | ||||||||||||||||
| Löslichkeit |
leicht löslich in unpolaren Lösungsmitteln, wie Tetrahydrofuran, Diethylether, n-Hexan, Benzol und Toluol[2], sowie in protischen Lösungsmitteln unter Protonierung | |||||||||||||||
| Sicherheitshinweise | ||||||||||||||||
| ||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | ||||||||||||||||



Vorkommen und Darstellung
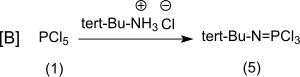
Die konvergente Synthese von P4-t-Bu[8] geht aus von Phosphorpentachlorid (1) und führt im Zweig [A] zunächst via [A1] über das nicht isolierte Chlor(dimethylamino) phosphoniumchlorid (2)[1] zum gut charakterisierbaren Aminotris(dimethylamino)phosphoniumtetrafluoroborat (3) und weiter via [A2] zum flüssigen Iminotris(dimethylamino)phosphoran[9] (4)
phosphoran.svg.png.webp)
und im Zweig [B] mit Phosphorpentachlorid und tert-Butylammoniumchlorid zum tert-Butylphosphorimid-trichlorid (5)[10]
Die Reaktion [C] von überschüssigem (4) mit (5) liefert mit 93%iger Ausbeute das Hydrochlorid des Zielprodukts P4-t-Bu (6),
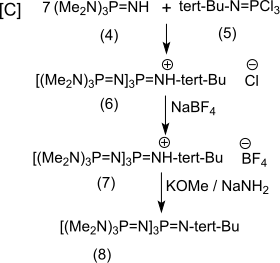
das ebenfalls in das Tetrafluoroboratsalz (7) überführt wird, aus dem die freie Base (8) mit Kaliummethanolat/Natriumamid[1] oder mit Kaliumamid in flüssigem Ammoniak[2] fast quantitativ gewonnen werden kann. Die Überführung der hygroskopischen und gut wasserlöslichen Hydrochloride und der flüssigen freien Basen in die in Wasser schwerlöslichen und festen Tetrafluoroborate erleichtert die Handhabung der Substanzen erheblich.
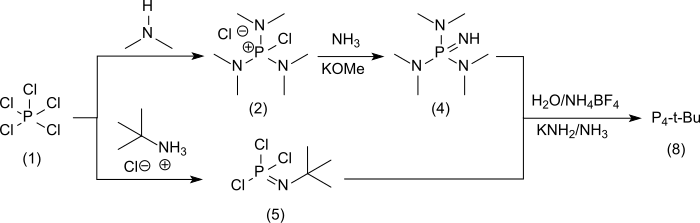
Die relativ unkomplizierte konvergente Synthese mit einfach zugänglichen Reaktanden und sehr guten Ausbeuten der Zwischenstufen machen P4-t-Bu zu einer interessanten Phosphazen-Superbase.[11]
Eigenschaften
P4-t-Bu ist mit einem extrapolierten pKa-Wert von 42.1 in Acetonitril eine der stärksten neutralen Stickstoffbasen und im Vergleich zu der starken Base DBU mit einem pKa-Wert von 24.3 um 18 Größenordnungen stärker basisch.[1] Die Verbindung ist sehr gut in unpolaren Lösungsmitteln, wie z. B. Hexan, Toluol oder Tetrahydrofuran löslich und ist meist als 0,8 bis 1 molare Lösung in Hexan im Handel.[11] Protonierung bereits in schwach sauren Medien erzeugt das extrem delokalisierte und weiche Kation P4-t-Bu-H-Kation und bewirkt neben einem sehr starken Solubilisierungseffekt auch eine extreme Beschleunigung von Additionsreaktionen bereits bei Temperaturen unter −78 °C.
Die außerordentlich hohe Basizität bei geringer Nukleophilie verdankt P4-t-Bu seiner sehr hohen sterischen Hinderung und der Beteiligung vieler Donorgruppen an der Konjugation in der räumlich anspruchsvollen Struktur des durch Protonierung gebildeten Kations.
Die Base P4-t-Bu ist ein extrem hygroskopischer Feststoff, der bis 120 °C thermisch und gegen (trockenen) Sauerstoff und Basen chemisch stabil ist.[2] Spuren von Wasser und protischen Verunreinigungen können durch Zugabe von Bromethan beseitigt werden. Die Base ist sowohl sehr hydrophil, als auch sehr lipophil und lässt sich über die Bildung des schwerlöslichen Tetrafluoroboratsalzes leicht und nahezu vollständig aus Reaktionsgemischen wiedergewinnen.
Wegen seiner äußerst schwachen Lewis-Basizität unterdrückt das Kation von P4-t-Bu typische Nebenreaktionen von Metallorganylen, wie z. B. Aldolkondensationen, wie sie durch Lithiumamide wie Lithiumdiisopropylamid (LDA) verursacht werden können.[12]
Anwendungen
Die neutrale Superbase P4-t-Bu ist ionischen Basen überlegen, wenn diese empfindlich sind gegen Oxidation oder Nebenreaktionen, wie z. B. Acylierung, Löslichkeitsprobleme verursachen oder Lewis-Säure-katalysierte Nebenreaktionen bewirken, wie z. B. Aldolreaktionen, Epoxid-Ringöffnungen usw.
Die Dehydrohalogenierung von n-Alkylbromiden, wie z. B. von 1-Bromoctan mit P4-t-Bu liefert 1-Octen in fast quantitativer Ausbeute (96 %) unter milden Bedingungen gegenüber dem System Kalium-tert-butanolat/18-Krone-6 mit lediglich 75 % Ausbeute.[13]
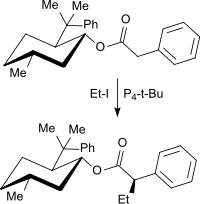
Alkylierungsreaktionen an schwach aciden Methylengruppen, z. B. bei Carbonsäureestern oder Nitrilen, verlaufen mit hoher Ausbeute und Selektivität. So wird bei der Umsetzung von 8-Phenylmenthylphenylacetat mit Iodethan in Gegenwart von P4-t-Bu ausschließlich das Monoethylderivat in der Z-Konfiguration (95 %) in 95%iger Ausbeute erhalten.[14]
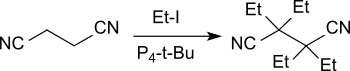
1,2-Ethandinitril reagiert mit Iodethan in Gegenwart von P4-t-Bu in 98%iger Ausbeute zum Tetraethylderivat, ohne dass es dabei zur Thorpe-Ziegler-Reaktion unter Bildung eines cyclischen α-Ketonitrils kommt.[2]
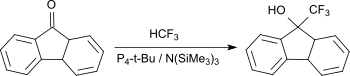
Trifluormethylierung von Ketonen, wie z. B. Benzophenon gelingt auch mit dem sehr reaktionsträgen Fluoroform (HFC-23) in Gegenwart von P4-t-Bu und Tris(trimethylsilyl)amin bei Raumtemperatur in guten Ausbeuten bis 84 %.[15]
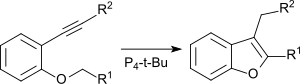
Intramolekulare Cyclisierung von ortho-Alkinylphenylethern führt in Gegenwart von P4-t-Bu unter milden Bedingungen ohne Metallkatalysatoren zu substituierten Benzofuranen.[16]
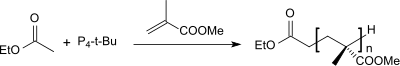
Die extreme Basizität von P4-t-Bu legte bereits früh die Vermutung nahe, dass sich diese Superbase als Initiator für die anionische Polymerisation eignen sollte. Aus Methylmethacrylat mit dem Ethylacetat/P4-t-Bu-Initiatorsystem konnte im Lösungsmittel THF Polymethylmethacrylat (PMMA) mit enger Polydispersität und Molmassen bis 40,000 g·mol−1 erhalten werden.[12]
Anionische Polymerisation von Ethylenoxid mit dem Initiatorsystem n-Butyllithium/P4-t-Bu liefert definierte Polyethylenoxide mit niedriger Polydispersität.[17]
Cyclische Siloxane, wie z. B. Hexamethylcyclotrisiloxan oder Decamethylcyclopentasiloxan können ebenfalls mit katalytischen Mengen von P4-t-Bu und Wasser oder Silanolen als Initiator unter guter Molmassenkontrolle zu thermisch sehr stabilen Polysiloxanen mit Zerfallstemperaturen > 450 °C polymerisiert werden.[18][5] Wegen seiner extremen Basizität absorbiert P4-t-Bu begierig Wasser und Kohlendioxid, die jedoch beide die anionische Polymerisation inhibieren. Erhitzen auf Temperaturen > 100 °C zur Entfernung von CO2 und Wasser setzt die anionische Polymerisation wieder in Gang.
Die extreme Hygroskopie der Phosphazenbase P4-t-Bu als Substanz und in Lösungen erfordert enormen Aufwand bei Lagerung und Handhabung und steht ihrer breiteren Verwendung entgegen.
Einzelnachweise
- R. Schwesinger et al.: Extremely strong, uncharged auxiliary bases; Monomeric and polymer-supported polyaminophosphazenes (P2-P5). In: Liebigs Ann. Chem. Band 7, 1996, S. 1055–10081, doi:10.1002/jlac.199619960705.
- R. Schwesinger, Y. Kondo: Phosphazene Base P4-t-Bu. In: e-EROS Encyclopedia of Reagents for Organic Synthesis. 2010, doi:10.1002/047084289X.rp150.pub2.
- Datenblatt Phosphazene base P4-t-Bu solution, ~0.8 M in hexane bei Sigma-Aldrich, abgerufen am 29. Dezember 2016 (PDF).
- R. Schwesinger et al.: Wie stark und wie gehindert können ungeladene Phosphazene sein? In: Angew. Chem. Band 105, Nr. 9, 1993, S. 1420–1422, doi:10.1002/ange.19931050940.
- Patent US6353075B1: Polymerization of siloxanes. Angemeldet am 9. Dezember 1999, veröffentlicht am 5. März 2002, Anmelder: Dow Corning Ltd., Erfinder: P. Hupfield, A. Surgenor, R. Taylor.
- J. Saame et al.: Experimental basicities of superbasic phosphonium ylides and phosphazenes. In: J. Org. Chem. Band 81, Nr. 17, 2016, S. 7349–7361, doi:10.1021/acs.joc.6b00872.
- E.D. Nacsa, T.H. Lambert: Higher-order cyclopropenimine superbases. Direct neutral Bronsted base catalyzed Michael reactions with α-aryl esters. In: J. Am. Chem. Soc. Band 137, Nr. 32, 2015, S. 10246–10253, doi:10.1021/jacs.5b05033.
- V. Gupta: New synthetic methods for biologically active aromatic heterocycles. Hrsg.: Iowa State University. Ames, Iowa 2010 (Online).
- Patent EP0921128B1: Process of preparing iminotris(dimethylamino)phosphorane. Angemeldet am 3. Dezember 1998, veröffentlicht am 25. September 2002, Anmelder: Mitsui Chemicals, Inc., Erfinder: T. Nobori et al..
- R. Schwesinger, J. Willaredt, H. Schlemper, M. Keller, D. Schmitt, H. Fritz: Novel, Very Strong, Uncharged Auxiliary Bases; Design and Synthesis of Monomeric and Polymer-Bound Triaminoiminophosphorane Bases of Broadly Varied Steric Demand. In: Chem. Ber. Band 127, Nr. 12, 1994, S. 2435–2454, doi:10.1002/cber.199441271215.
- Strong and Hindered Bases in Organic Syntheses. (PDF; 1,2 MB) In: sigmaaldrich.com. Sigma-Aldrich, abgerufen am 20. Dezember 2016 (englisch).
- T. Pietzonka, D. Seebach: Die P4-Phosphazenbase als Teil eines metallfreien Initiatorsystems für die anionische Polymerisation von Methacrylsäuremethylester. In: Angew. Chem. Band 105, Nr. 5, 1993, S. 741–742, doi:10.1002/ange.19931050514.
- R. Schwesinger, H. Schlemper: Peralkylierte Polyaminophosphazene – extrem starke neutrale Stickstoffbasen. In: Angew. Chem. Band 99, Nr. 11, 1987, S. 1212–1214, doi:10.1002/ange.19870991134.
- A. Solladié-Cavallo, A.G. Csaky, I. Gantz, J. Suffert: Diastereoselective Alkylation of 8-Phenylmenthyl Phenylacetate: Aggregated Lithium Enolate versus "Naked" Enolate. In: J. Org. Chem. Band 59, Nr. 18, 1994, S. 5343–5346, doi:10.1021/jo00097a041.
- S. Okusu, K. Hirano, E. Tokunaga, N. Shibata: Organocatalyzed trifluormethylation of ketones and sulfonyl fluorides by fluoroform under a superbase system. In: ChemistryOpen. Band 4, 2015, S. 581–585, doi:10.1002/open.201500160.
- C. Kanazawa, K. Goto, M. Terada: Phosphazene base-catalyzed intramolecular cyclization for efficient synthesis of benzofurans via carbon-carbon bond formation. In: Chem. Commun. 2009, S. 5248–5250, doi:10.1039/B913588J.
- B. Eßwein, M. Möller: Polymerisation von Ethylenoxid mit Alkyllithiumverbindungen und der Phosphazenbase "t Bu-P4". In: Angew. Chem. Band 108, Nr. 6, 1996, S. 703–705, doi:10.1002/ange.19961080620.
- P.C. Hupfield, R.G. Taylor: Ring-opening polymerisation of siloxanes using phosphazene base catalysts. In: J. Inorg. Organomet. Polym. Mater. Band 9, Nr. 1, 1999, S. 17–34, doi:10.1023/A:1021429320083.