Entropie
Die Entropie (Kunstwort altgriechisch ἐντροπία entropía, von ἐν en ‚an‘, ‚in‘ und τροπή tropḗ ‚Wendung‘) ist eine fundamentale thermodynamische Zustandsgröße mit der SI-Einheit Joule pro Kelvin (J/K).
| Physikalische Größe | |||||||
|---|---|---|---|---|---|---|---|
| Name | Entropie | ||||||
| Formelzeichen | |||||||
| |||||||

Die Entropie eines Systems steigt bei jedem spontan darin ablaufenden Prozess sowie bei Zufuhr von Wärme oder Materie. Spontan ablaufende Prozesse sind z. B. Vermischung, Wärmeleitung, chemische Reaktion, aber auch Umwandlung von mechanischer Arbeit in Innere Energie durch Reibung (siehe Dissipation, Energieentwertung). Abnehmen kann die Entropie eines Systems nur durch Abgabe von Wärme oder Materie. Daher kann in einem abgeschlossenen System (einem System, bei dem es keinen Energie- oder Materieaustausch mit der Umgebung gibt) die Entropie nicht abnehmen, sondern im Laufe der Zeit nur gleich bleiben oder zunehmen (Zweiter Hauptsatz der Thermodynamik). Hat ein System die maximal mögliche Entropie erreicht, kommen alle spontan darin ablaufenden Prozesse zum Erliegen, und das System ist in einem stabilen Gleichgewichtszustand.
Beispielsweise beobachten wir, dass in einem System aus einem kalten und einem heißen Körper in einer Isolierbox, d. h. in einem praktisch abgeschlossenen System, Wärmetransport einsetzt und der Temperaturunterschied verschwindet. Beide Körper werden nach einer gewissen Zeit die gleiche Temperatur haben, womit das System den Zustand größter Entropie erreicht hat und sich nicht weiter verändert. Wir beobachten in einem solchen geschlossenen System praktisch niemals das spontane Abkühlen des kälteren Körpers und das Erhitzen des wärmeren.[Anm. 1]
Spontane Prozesse, bei denen die Entropie in einem abgeschlossenen System zunimmt, können ohne äußeren Eingriff nicht in umgekehrter zeitlicher Richtung ablaufen, sie werden als irreversibel bezeichnet. Um ein System nach einem irreversiblen Prozess wieder in den Ausgangszustand zu bringen, muss es mit seiner Umgebung gekoppelt werden, die den Zuwachs an Entropie aufnimmt und ihren eigenen Zustand dadurch auch verändert.
In der statistischen Mechanik wird durch makroskopische thermodynamische Größen ein Makrozustand eines Systems definiert, der durch eine hohe Anzahl verschiedener Mikrozustände realisiert sein kann. Diese Mikrozustände können durch innere Prozesse ineinander übergehen. Ihre Anzahl bestimmt die Entropie, die das System in diesem gegebenen Makrozustand besitzt. In einem System, das in einem beliebigen Anfangszustand ist und sich selbst überlassen wird, bewirken dann die spontan ablaufenden inneren Vorgänge, dass der Zustand des Systems sich mit größter Wahrscheinlichkeit demjenigen Makrozustand annähert, der bei gleicher Energie durch die größte Anzahl verschiedener Mikrozustände realisiert sein kann, also die höchstmögliche Entropie hat. Dieser Vorgang bewirkt die Annäherung an den (makroskopischen) Gleichgewichtszustand (Relaxation).
Dies wird umgangssprachlich häufig dadurch umschrieben, dass Entropie ein „Maß für Unordnung“ sei. Allerdings ist Unordnung kein definierter physikalischer Begriff und hat daher kein physikalisches Maß. Richtiger ist es, man begreift die Entropie als ein wohldefiniertes objektives Maß für die Menge an Information, die benötigt würde, um von einem beobachtbaren Makrozustand auf den tatsächlich vorliegenden Mikrozustand des Systems schließen zu können. Dies ist gemeint, wenn die Entropie auch als „Maß für die Unkenntnis der Zustände aller einzelnen Teilchen“ umschrieben wird.[1]
Geschichtlicher Überblick
In der Geschichte der Physik gab es lange Zeit eine Auseinandersetzung über die Bedeutung von Wärme: Die eine Seite vertrat die Theorie, dass die Wärmeerscheinungen allein in der vis viva („lebendige Kraft“ = kinetische Energie) der Atome begründet seien; die andere behauptete, dass die Wärme eine Substanz sei, und gab ihr den Namen Caloricum (französisch calorique, englisch caloric).
Antoine Laurent de Lavoisier unterschied 1789 chaleur (Wärme) von calorique (Caloricum). Das Caloricum sollte unter anderem eine abstoßende Kraft zwischen den Atomen eines Festkörpers bewirken, so dass dieser bei Zufuhr einer ausreichenden Menge von Caloricum erst flüssig und dann gasförmig würde.[2] Lavoisier wollte sich aber nicht festlegen, ob die vis viva oder die Caloricum-Substanz die Ursache für die Wärmeerscheinungen ist.[3] Joseph Black unterschied temperature von der quantity of heat, u. a. anhand der latenten Wärme beim Schmelzen. Er bemerkte, dass die Wärmemenge mit dem aus einem Kessel entweichenden Dampf mittransportiert werden müsse.[4][5]
Benjamin Thompson, Reichsgraf von Rumford, untersuchte in seiner Münchner Zeit 1798 die Temperatur von Spänen, die beim Bohren von Kanonenrohren entstehen. Aufgrund der beliebig großen Wärmemenge, die dabei aus der mechanischen Bohrarbeit entstehen konnte, zweifelte er daran, dass das Caloricum eine (erhaltene) Substanz sein könnte, wodurch er den Vertretern der vis-viva-Theorie Auftrieb gab.[6][5]
Der Namensgeber des Carnot-Prozesses, Nicolas Léonard Sadi Carnot, schrieb 1824[7], dass die Kraft einer Dampfmaschine nicht am Verbrauch von calorique liegt, sondern an ihrem Transport von einem warmen Körper auf einen kalten, und bereitete damit den Entropiebegriff vor. Mit den Experimenten von Robert Mayer und James Prescott Joule wurde Anfang der 1840er Jahre gezeigt, dass die mechanisch erzeugte Wärme in einem festen Verhältnis zur aufgewendeten mechanischen Arbeit steht. Dies war die Grundlage für den 1847 von Hermann von Helmholtz allgemein formulierten Energieerhaltungssatz, also den ersten Hauptsatz der Thermodynamik. Seitdem hat der physikalische Begriff Wärme die Bedeutung einer Energie.
Weitere 20 Jahre später stellte dann Rudolf Clausius fest, dass bei einer Übertragung der Energieform Wärme auch eine zweite mengenartige Größe fließen muss. Diese Größe, die er auch quantitativ definierte, sah er als die Ursache für die „Disgregation“ eines festen Körpers beim Schmelzen an und nannte sie Entropie.[8][9] Wie von Wilhelm Ostwald 1908 sowie Hugh Longbourne Callendar 1911 herausgearbeitet, entspricht die Entropie bei Clausius dem calorique bei Lavoisier und Carnot.[10][11][12]
Mit Arbeiten von Ludwig Boltzmann und Willard Gibbs gelang es um 1875, der Entropie eine statistische Definition zu geben, welche die von Clausius makroskopisch definierte Größe mikroskopisch erklärt.[13] Die Entropie eines Makrozustands wird dabei durch die Wahrscheinlichkeiten der Mikrozustände berechnet:



Der Proportionalitätsfaktor wurde nach seinem Tod als Boltzmann-Konstante bezeichnet, Boltzmann selbst hat ihren Wert jedoch nie bestimmt.

Die so auf statistische Weise definierte Entropie kann in vielen Kontexten sinnvoll verwendet werden.
Zusammenhänge zwischen Entropie und Information ergaben sich schon im 19. Jahrhundert durch die Diskussion über den maxwellschen Dämon, ein Gedankenexperiment, das im Rahmen der Miniaturisierung im Computerzeitalter wieder aktuell wurde. Die Informationstheorie nutzt die Shannonsche Informationsentropie, die der statistischen Interpretation entspricht, als abstraktes Maß der Information ohne direkten Bezug zur physikalischen Realisation. Auch Norbert Wiener nutzte den Begriff der Entropie zur Beschreibung von Informationsphänomenen, allerdings mit entgegengesetztem Vorzeichen. Dass sich die Konvention von Shannon durchgesetzt hat, ist vor allem der besseren technischen Verwertbarkeit seiner Arbeiten zuzuschreiben.[14]
Klassische Thermodynamik
In der Thermodynamik kann ein System auf zwei Arten mit seiner Umgebung Energie austauschen: als Wärme oder als Arbeit, wobei je nach System und Prozessführung verschiedene Varianten der Arbeit existieren, u. a. Volumenarbeit, Hubarbeit und magnetische Arbeit. Im Zuge eines solchen Energieaustauschs kann sich die Entropie sowohl des Systems als auch der Umgebung ändern. Nur wenn die Summe aller Entropieänderungen positiv ist, erfolgt die Änderung spontan.
Grundlagen
Die Entropie (Einheit J/K) ist eine extensive Zustandsgröße eines physikalischen Systems und verhält sich bei Vereinigung mehrerer Systeme additiv wie auch das Volumen, die elektrische Ladung oder die Stoffmenge. Der Physiker Rudolf Clausius führte diesen Begriff 1865 zur Beschreibung von Kreisprozessen ein. Dividiert man durch die Masse des Systems, erhält man die spezifische Entropie mit der Einheit J/(kg·K) als intensive Zustandsgröße.

Das Differential ist nach Clausius bei reversiblen Vorgängen zwischen Systemen im Gleichgewicht das Verhältnis von übertragener Wärme und absoluter Temperatur :




Diese Entropieänderung ist bei Wärmezufuhr positiv, bei Wärmeabfuhr negativ. In dieser Schreibweise wird bei ein nicht-kursives benutzt, um hervorzuheben, dass es sich um ein vollständiges Differential handelt, im Gegensatz zu , das kein vollständiges Differential sein kann, weil eine Prozessgröße ist. In diesem Zusammenhang spielt also die reziproke absolute Temperatur die Rolle eines „integrierenden Bewertungsfaktors“, der aus der reversibel zu- oder abgeführten Wärme, einem – mathematisch gesehen – unvollständigen Differential, ein zugehöriges vollständiges Differential macht.[15] Dadurch ist die Änderung der Entropie bei reversiblen Prozessen – im Gegensatz zur zu- oder abgeführten Wärme – wegunabhängig. Mit der Festlegung eines beliebigen Werts für einen Bezugszustand wird die Entropie damit zu einer allein durch den jeweiligen Zustand gegebenen Zustandsgröße.



Insofern kann man die Entropie bei reversibler Prozessführung auch als die „mit bewertete Wärmeenergie“ definieren. Weiter unten wird das Problem behandelt, wie weit die Energie eines Systems in Arbeit umgewandelt werden kann.

Benutzt man den ersten Hauptsatz der Thermodynamik, , also dass die Energieänderung sich zusammensetzt aus zugeführter Arbeit und Wärme, und setzt für die Arbeit alle dem Experimentator mittels Änderung der Systemgrößen möglichen Prozesse, erhält man aus (1) für die Änderung der Entropie als Funktion der thermodynamischen Variablen (immer noch im reversiblen Fall)




Clausius behandelte auch irreversible Prozesse und zeigte, dass in einem isolierten thermodynamischen System die Entropie dabei nie abnehmen kann:

wobei das Gleichheitszeichen nur bei reversiblen Prozessen gilt. ist die Entropieänderung des Systems mit für die Entropie des Zustandes am Anfang der Zustandsänderung und für den Zustand am Ende des Prozesses.



Aus (2) folgt für geschlossene Systeme, bei denen Wärmeenergie die Systemgrenzen passieren kann, die Ungleichung:

ist der Entropie-Anteil, der sich aus der Zufuhr von Wärme über die Systemgrenze hinweg ergibt. Die Formel gilt auch für die Abfuhr von Wärme aus dem System, in diesem Falle ist negativ. Die Ungleichung (3a) wird nur für rein reversible Prozesse zu einer Gleichung.

Bei der Analyse thermodynamischer Systeme in der Technik führt man oft eine Bilanzanalyse durch. Hierzu schreibt man die Ungleichung (3a) in der folgenden Form:[16]

Dabei ist der Entropieanteil der durch irreversible Vorgänge im Inneren des Systems entsteht. Hierzu gehören etwa Vermischungsvorgänge nach dem Entfernen einer inneren Trennwand, thermische Ausgleichsvorgänge, die Umwandlung von elektrischer oder mechanischer Energie (ohmscher Widerstand, Rührwerk) in Wärme und chemische Reaktionen. Beschränken sich die irreversiblen Vorgänge ausschließlich auf die Dissipation von mechanischer oder elektrischer Arbeit , dann kann durch die Arbeit bzw. die dissipierte Leistung ausgedrückt werden.





Läuft der irreversible Prozess quasistatisch ab, so dass das System immer nahe einem Gleichgewichtszustand ist, so kann (3) auch mit zeitlichen Ableitungen geschrieben werden.

Dabei wird als Entropietransportstrom und als Entropieproduktionsstrom bezeichnet.[16]


Aus dem ersten Hauptsatz der Thermodynamik

folgt, dass das Produkt den nicht verwerteten Anteil („Abwärme“) bei der isothermen Erzeugung von Arbeit aus vorhandener innerer Energie darstellt. Der Maximalwert dieser Arbeit ist die sogenannte freie Energie



- .

Dies ist eine äquivalente Form des 2. Hauptsatzes.
Eine Konsequenz dessen ist die Unmöglichkeit eines Perpetuum Mobile 2. Art. Clausius formulierte:
„Es existiert kein Kreisprozess, dessen einzige Wirkung darin besteht, Wärme von einem kälteren Reservoir zu einem wärmeren Reservoir zu transportieren.“
Offenbar hätte man sonst eine unerschöpfliche Energiequelle konstruiert. Wäre es nämlich möglich, einen solchen Kreisprozess zu konstruieren, könnte man dem warmen Reservoir unentwegt Energie entnehmen und damit Arbeit verrichten. Die dissipierte Arbeit würde dann dem kalten Reservoir zugeführt und käme über den erwähnten Kreisprozess wieder dem warmen Reservoir zugute. Äquivalent dazu ist die Formulierung von William Thomson, dem späteren Lord Kelvin:
„Es existiert kein Kreisprozess, der eine Wärmemenge aus einem Reservoir entnimmt und vollständig in Arbeit verwandelt.“
Ein idealer, jederzeit umkehrbarer Prozess ohne Reibungsverluste wird auch reversibel genannt. Oft bleibt die Entropie während eines Prozesses unverändert, , bekanntes Beispiel ist die adiabate Kompression und Expansion im Zyklus einer Carnot-Maschine. Man nennt Zustandsänderungen mit konstanter Entropie auch isentrop, allerdings sind nicht alle isentropen Zustandsänderungen adiabatisch. Ist ein Prozess adiabatisch und reversibel, folgt jedoch stets, dass er auch isentrop ist.

Wird in einem Kreisprozess bei der Temperatur die Wärme aufgenommen und die Wärmemenge bei wieder abgegeben und erfolgen Wärmeaufnahme und Abgabe reversibel, so gilt, dass sich die Entropie dabei nicht ändert:




- ; oder .


Daraus lassen sich die maximale verrichtete Arbeit und der maximale Wirkungsgrad , der so genannte Carnot-Wirkungsgrad, ableiten:



Der carnotsche Wirkungsgrad stellt für alle Wärmekraftmaschinen das Maximum an Arbeitsausbeute dar. Reale Maschinen haben meistens einen erheblich geringeren Wirkungsgrad. Bei ihnen wird ein Teil der theoretisch verfügbaren Arbeit dissipiert, z. B. durch Reibung. Folglich entsteht in einer realen Maschine Entropie und es wird mehr Wärme an das kalte Reservoir abgeführt als notwendig. Sie arbeitet also irreversibel.
Der dritte Hauptsatz (der so genannte „Nernstsche Wärmesatz“) legt die Entropie einer perfekt kristallinen Substanz, bei der beispielsweise keine Spinentartung auftritt, am absoluten Nullpunkt als Null fest:

Eine Folgerung ist beispielsweise, dass die Wärmekapazität eines Systems bei tiefen Temperaturen verschwindet, und vor allem, dass der absolute Temperaturnullpunkt nicht erreichbar ist (das gilt auch bei Spinentartung).
Erfüllt eine Substanz nicht die Bedingung perfekt kristallin (wenn z. B. mehrere Konfigurationen vorliegen oder es sich um ein Glas handelt), kann ihr auch am absoluten Nullpunkt eine Entropie zugeschrieben werden (Nullpunktsentropie).
Partielle Ableitungen der Entropie
Aus dem 2. Hauptsatz folgen Aussagen über die partiellen Ableitungen der Entropie, z. B. nach der Temperatur oder dem Volumen . Mit dem zweiten Hauptsatz gilt zunächst, dass bei reversiblen Zustandsänderungen ist. Zusammen mit dem ersten Hauptsatz folgt daraus , weil nach dem ersten Hauptsatz für die Innere Energie gilt, dass die Summe der dem betrachteten System zugeführten Arbeit und der zugeführten Wärme (einzeln keine Zustandsfunktionen!) eine Zustandsfunktion ergibt, eben die „Innere Energie“ des Systems. Dabei wurde vorausgesetzt, dass die Änderungen von Volumen und Temperatur adiabatisch-langsam erfolgen, sodass keine irreversiblen Prozesse erzeugt werden.





Also

wobei eingesetzt wurde.

- bzw.
- .


Ähnliche Beziehungen ergeben sich, wenn das System außer von der Dichte bzw. vom Volumen noch von weiteren Variablen abhängt, z. B. von elektrischen oder magnetischen Momenten.
Aus dem 3. Hauptsatz folgt, dass sowohl als auch für verschwinden müssen, und zwar hinreichend rasch, was (wie man zeigen kann) nur erfüllt ist, wenn für tiefe Temperaturen nicht die klassische Physik, sondern die Quantenphysik gilt.



Statistische Physik
In der u. a. von James Maxwell begründeten statistischen Mechanik wird das Verhalten makroskopischer thermodynamischer Systeme durch das mikroskopische Verhalten seiner Komponenten, also Elementarteilchen und daraus zusammengesetzter Systeme wie Atome und Moleküle, erklärt. Bezüglich der Entropie stellt sich die Frage, wie sie hier gedeutet werden kann, und ob der zeitlich gerichtete zweite Hauptsatz aus einer mikroskopischen zeitumkehrinvarianten Theorie hergeleitet werden kann.
Ein Mikrozustand ist klassisch gegeben durch Angabe aller Orte und Impulse der zum System zählenden Teilchen. Ein solcher Mikrozustand ist demnach ein Punkt in einem 6N-dimensionalen Raum, der in diesem Zusammenhang Phasenraum genannt wird. Die kanonischen Gleichungen der klassischen Mechanik beschreiben die zeitliche Evolution des Systems, die Phasentrajektorie. Alle unter gegebenen makroskopischen Randbedingungen, wie z. B. Gesamtenergie , Volumen und Teilchenzahl , erreichbaren Phasenpunkte bilden ein zusammenhängendes Phasenraumvolumen .




Um 1880 konnte Ludwig Boltzmann auf mikroskopischer Ebene eine Größe finden, die die Definition der thermodynamischen Entropie erfüllt:

Die Konstante ist die Boltzmannkonstante. Die Entropie ist also proportional zum Logarithmus des zu den Werten der thermodynamischen Variablen gehörigen Phasenraumvolumens.
Eine äquivalente Formel lautet

(mit der Wahrscheinlichkeit für den Mikrozustand bei Kenntnis der zum thermodynamischen System gehörigen Variablen, und dem Integral über den Phasenraum). Sie führt – wenn man mit das natürliche Maß auf dem Phasenraum benutzt und die Wahrscheinlichkeit in Abwesenheit anderer Information als konstant ansieht ( mit dem Phasenraumvolumen , das zu den Werten der thermodynamischen Variablen gehört) – sofort auf die „Boltzmann-Formel“ (da die Integration unabhängig von ist: und ). Ihre Ähnlichkeit mit Shannons Ausdruck für die Information legt nahe, die Entropie als das Informationsdefizit über den Mikrozustand zu interpretieren, das mit der Kenntnis der makroskopischen Variablen verbunden ist. Je größer die Entropie ist, desto weniger weiß man über den mikroskopischen Zustand und desto weniger Informationen sind über das System bekannt.





Letzteres wurde von E.T. Jaynes unter dem Stichwort „informationstheoretische Entropie“ zu einem Konzept ausgearbeitet, die Entropie als epistemische (er nannte es „anthropomorph“) Größe zu verstehen, etwa in folgendem Zitat:
“For example, I have been asked several times whether, in my opinion, a biological system, say a cat, which converts inanimate food into a highly organized structure and behavior, represents a violation of the second law. The answer I always give is that, until we specify the set of parameters which define the thermodynamic state of the cat, no definite question has been asked!”
„Zum Beispiel bin ich mehrmals gefragt worden, ob meiner Meinung nach ein biologisches System, etwa eine Katze, die unbelebte Nahrung in hoch organisierte Struktur und Verhalten verwandelt, eine Verletzung des zweiten Hauptsatzes darstellt. Die Antwort, die ich immer gebe, ist, dass, solange wir den Satz an Parametern, die den thermodynamischen Zustand der Katze festlegen, nicht spezifizieren, keine eindeutige Frage gestellt wurde.“
Es wird deutlich, dass die Entropie – wie überhaupt ein thermodynamisches System – erst durch eine Auswahl an Variablen definiert und von diesen abhängig ist. Einem Mikrozustand kann sie nicht zugeordnet werden. Kritisiert wird daran, dass hier die Entropie den Rang einer subjektiven Größe zu haben scheint, was in einer objektiven Naturbeschreibung nicht angemessen ist.
Beweis des zweiten Hauptsatzes
Vor allem Boltzmann versuchte, den 2. Hauptsatz (dass die Entropie nur zunehmen kann) statistisch herzuleiten. Die anschauliche Vorstellung ist die, dass etwa bei einem Mischungsvorgang etwas sehr Wahrscheinliches passiert, während der umgekehrte Vorgang einer Entmischung sehr unwahrscheinlich wäre. Dies galt es mathematisch zu präzisieren, mit seinem H-Theorem hatte er hier einen Teilerfolg. Allerdings wird mit dem „Loschmidt'schen Umkehreinwand“ verdeutlicht, dass mikroskopisch jeder Vorgang genauso rückwärts laufen könnte und daher ein zeitgerichtetes Gesetz mikroskopisch grundsätzlich nicht hergeleitet werden kann. Auch der Wiederkehrsatz stellt die Möglichkeit eines solchen Gesetzes in Frage.
Im informationstheoretischen Konzept verstanden bedeutet der 2. Hauptsatz, dass die Information über den Mikrozustand bei Beobachtung der makroskopischen Variablen nur abnehmen kann. Hier ist der Beweis viel einfacher möglich:
Nach dem Satz von Liouville bleibt das Phasenraumvolumen der mit einem Anfangswert der thermodynamischen Variablen verbundenen Mikrozustände bei der Zeitentwicklung konstant.[18] Wenn man noch voraussetzt, dass die Beschreibung durch die thermodynamischen Variablen eindeutig ist, das heißt alle Mikrozustände makroskopisch gesehen beim selben Endzustand landen, kann also das Phasenraumvolumen der mit diesem Endwert der thermodynamischen Variablen verbundenen Mikrozustände nicht kleiner sein als das anfängliche Phasenraumvolumen. Es kann aber größer sein, da nicht notwendigerweise alle Mikrozustände „angesteuert“ werden. Die Entropie kann also nur zunehmen.
Man kann das anders formulieren. Man unterscheidet zwischen Von-Neumann- oder „fine-grained“- oder „entanglement“-Entropie (also der von Mikrophysik, d. h. wellenmechanisch korrelierten Systemen) und thermischer Entropie (also der Entropie in klassischer, makroskopischer Thermodynamik, auch genannt „coarse-grained“-Entropie). Ohne Korrelation ist die Entanglement-Entropie () null (jeweils nur ein Zustand, , der „reine Zustand“). Mit entanglement (Korrelation) stehen mehr Zustände zur Verfügung, und die Entanglement-Entropie ist größer als null. In der Makrophysik betrachtet man Phasenraumbereiche wie das Kugelvolumen („coarse-graining“) um einen Punkt, also nicht einzelne Punkte bzw. Mikrozustände. Der mit Anfangsbedingungen definierte Bereich des Phasenraums eines Systems wird demzufolge von Kugelvolumen bedeckt, die mehr Phasenraumpunkte umfassen als im mikroskopischen Anfangszustand. Damit ist die Fine-grained-Entropie immer kleiner als die Coarse-grained-Entropie. Dies ist die Aussage des 2. Hauptsatzes. Als Information bezeichnet man die Differenz zwischen Coarse-grained-Entropie und Fine-grained-Entropie. Details findet man im Buch von Susskind und Lindesay.[19]


Die zeitliche Asymmetrie des zweiten Hauptsatzes betrifft hier also die Kenntnis des Systems, nicht die Ontologie des Systems selbst. Dadurch werden die Schwierigkeiten, aus einer bezüglich Zeitumkehr symmetrischen Theorie ein asymmetrisches Gesetz zu erhalten, vermieden. Allerdings geht in den Beweis auch die Eindeutigkeit der thermodynamischen Beschreibung ein, die auf stochastischen Argumenten beruht. Um die zeitliche Asymmetrie des Weltgeschehens zu verstehen, ist weiterhin ein Bezug zum Anfangszustand des Universums nötig.
Entropie als „Maß der Unordnung“


Eine anschauliche, aber wissenschaftlich nicht exakte Deutung der Entropie ist es, sie als Maß der Unordnung zu verstehen, siehe z. B. das Chemiebuch von Holleman-Wiberg.[20] Insbesondere in der Fotografie des Bechers im Beispiel zur Mischentropie sieht das rechte Bild der Abbildung 1 der vollständig Durchmischung für die meisten Betrachter ordentlicher aus als das linke mit den Schlieren, weshalb es dann unverständlich erscheint, es als den unordentlicheren Zustand mit höherer Entropie zu bezeichnen.
Diese Definition lässt sich allerdings anhand der schematischen Abbildung 2[21] mit den bisherigen Definitionen in Einklang bringen. Abbildung 2 stellt die 16 Zustände in einem Behälter dar, in dem sich jedes der vier enthaltenen Atome entweder auf der rechten oder auf der linken Seite des Behälters befinden kann. Die erste Spalte enthält den einen Zustand mit Atomen links, die zweite die vier Zustände mit Atomen links usw. Wenn man annimmt, dass alle 16 Zustände gleich wahrscheinlich sind, sind die Wahrscheinlichkeiten, für die einzelnen Spalten gerade mit gegeben, wobei das aus Abbildung 2 gerade die Anzahl der Zustände in den jeweiligen Spalten bezeichnet.



Nehmen wir nun an, dass wir makroskopisch unterscheiden könnten, wie viele Atome sich auf der linken Seite befinden. Die Wahrscheinlichkeit, dass sich alle vier Atome auf der linken Seite befinden, wäre als Beispiel daher gerade , während die mittlere Spalte eine höhere Wahrscheinlichkeit von besitzt. Mit der Formel hat der makroskopische Zustand mit also die höchste Entropie.




In Abbildung 2 erkennt man nun aber auch deutlich, dass die erste und letzte Spalte ordentlicher sind als die dazwischenliegenden Fälle mit höherer Entropie. Es können nun folgende Feststellungen gemacht werden: Wenn alle 16 Zustände gleich wahrscheinlich sind und man mit dem Zustand beginnt, ist es sehr wahrscheinlich, bei der nächsten Betrachtung einen der Zustände höherer Entropie vorzufinden. Allerdings kann das System auch von der mittleren Spalte in die erste oder letzte übergehen; es ist nur unwahrscheinlicher, den Zustand vorzufinden als den Zustand mit . In diesem rein statistischen Sinne kann das System also auch spontan in einen Zustand niedrigerer Entropie wechseln, es ist nur unwahrscheinlicher als der Wechsel in Richtung höherer Entropie.
Dies ist immer noch der Fall, wenn man Atome in einem Behälter betrachtet. Die Möglichkeit, dass diese spontan in den Zustand niedrigerer Entropie wechseln, bei dem sich alle Atome links befinden, ist nicht auszuschließen, aber sehr unwahrscheinlich.

Entropie als mengenartige Größe
Für die Entropie als extensive Größe kann eine lokale Bilanzgleichung (Kontinuitätsgleichung) formuliert werden[22] [23]:

Hier ist die Dichte der Entropie, die Stromdichte und die Erzeugungsrate pro Volumen. In manchen Lehrwerken zur Physik, insbesondere denen des Karlsruher Physikkurses, wird eine Vorstellung der Entropie als eine extensive und „mengenartige Größe“ nahegelegt. Im Fall der Entropie ist das, was sie misst, nach Wilhelm Ostwald und Hugh Longbourne Callendar mit dem carnotschen Caloricum zu identifizieren.[10][11] Da das Caloricum weitgehend mit dem umgangssprachlichen Wärmebegriff übereinstimmt, kann die Entropie demnach auch als Maß für die (umgangssprachliche) Wärme verstanden werden.[24][25][26][27] Man beachte dabei, dass in der Kontinuitätsgleichung auf der rechten Seite eine Erzeugungsrate steht, weshalb aus ihr kein Erhaltungssatz abgeleitet werden kann.



Von zwei ansonsten gleichen Körpern enthält derjenige mehr Entropie, dessen Temperatur höher ist. Fasst man zwei Körper zu einem einzigen System zusammen, ist die Gesamtentropie die Summe der Entropien beider Körper.[12][28]
Stehen zwei Körper unterschiedlicher Temperatur miteinander in wärmeleitendem Kontakt, verursacht die Temperaturdifferenz einen Entropiestrom. Aus dem wärmeren Körper fließt Entropie ab, wodurch sich dessen Temperatur absenkt. Der kältere Körper nimmt diese Entropie (und die zusätzlich in diesem Prozess neu erzeugte Entropie) auf, wodurch seine Temperatur steigt. Der Prozess kommt zum Erliegen, wenn die Temperaturen beider Körper gleich geworden sind.
Als mengenartige Größe ist die Entropie zum Beispiel mit der elektrischen Ladung vergleichbar (für die allerdings ein strikter Erhaltungssatz gilt): Ein geladener Kondensator enthält elektrische Ladung, und damit auch elektrische Energie. Beim Entladevorgang fließt über den Stromkreis nicht nur elektrische Ladung von der einen Kondensatorplatte auf die andere, sondern es fließt auch Energie vom Kondensator zu einem Verbraucher, wo sie in andere Energieformen umgewandelt werden kann. Entsprechend wird bei der Wärmeübertragung von einem heißen auf einen kalten Körper außer der thermischen Energie noch eine andere mengenartige Größe übertragen: die Entropie. Genauso wie der Potentialunterschied zwischen den Kondensatorplatten – sprich: die elektrische Spannung – den elektrischen Strom antreibt, sorgt der Temperaturunterschied zwischen den beiden Reservoirs für einen Entropiestrom. Befindet sich zwischen den beiden Körpern eine Wärmekraftmaschine, kann ein Teil der Wärme in eine andere Energieform umgewandelt werden. Die Wärmeübertragung lässt sich also rein formal analog zu einem elektrischen Stromkreis beschreiben, wobei allerdings die während des Vorgangs neu erzeugte Entropie zusätzlich zu berücksichtigen ist.[12][25]
Eine Energieübertragung – also die Arbeit oder die Wärme – lässt sich als das Produkt aus der Änderung einer mengenartigen extensiven Größe mit einer konjugierten intensiven Größe schreiben. Beispiele für solche Paare sind die elektrische Ladung und das elektrische Potential oder das Volumen und der (negativ genommene) Druck eines Gases. Durch Zufuhr der extensiven Größe steigt (im Allgemeinen) die intensive Größe. Fügt man beispielsweise eine kleine Ladungsmenge einer Kondensatorplatte zu, die sich auf dem Potential befindet, verrichtet man an der Kondensatorplatte die Arbeit und erhöht damit das Potential der Platte. Entsprechendes gilt auch für andere Größenpaare. Für thermische Vorgänge entspricht nun die Temperatur dem Potential und die Entropie der Ladung: Wenn man einem Körper die Entropiemenge hinzufügt, wird die Wärme übertragen und es steigt (außer bei Phasenumwandlung) die Temperatur .[12]





Die Entropie ist zwar keine Erhaltungsgröße wie die elektrische Ladung, denn Entropie kann erzeugt werden. Nach dem zweiten Hauptsatz der Thermodynamik kann sie aber nicht vernichtet werden. Es gilt also . Solange in einem abgeschlossenen System die Entropie gleich bleibt , sind alle Vorgänge, die sich in dem System abspielen, umkehrbar (= reversibel). Sobald jedoch Entropie erzeugt wird , was z. B. durch Reibung, durch Erwärmung eines ohmschen Widerstandes oder durch Mischungsvorgänge geschehen kann, gibt es kein Zurück mehr, ohne dass durch äußeres Zutun Entropie abfließen kann. Man spricht dann von irreversiblen Vorgängen.



Eine weitere Besonderheit der Entropie ist, dass man einem Körper nicht beliebig viel Entropie entziehen kann. Während man der Platte eines Kondensators Ladung zuführen und wegnehmen kann, wodurch das Potential positiv oder negativ wird, gibt es für die Entropieentnahme eine natürliche Grenze, nämlich den absoluten Nullpunkt der Temperatur. Insbesondere kann die absolute Temperatur nie negativ werden.
Anwendungsbeispiele
Mischung von warmem und kaltem Wasser
Die Zunahme der Entropie wird an einem System, das mit der Umgebung weder Masse noch Energie austauscht (abgeschlossenes System), durch die Vermischung von zwei Wassermengen unterschiedlicher Temperatur gezeigt. Da es sich um einen isobaren Vorgang handelt, wird für die Energiebilanz die Zustandsgröße Enthalpie verwendet.
Zustandsgrößen für Wasser nach den Gleichungen aus: Properties of Water and Steam Industrie-Standard IAPWS-IF97
System10: Masse m10 = 1 kg, Druck = 1 bar, Temperatur = 10 °C, Enthalpie h10 = 42,12 kJ/kg, Entropie s10 = 151,1 J/kg K; System30: Masse m30 = 1 kg, Druck = 1 bar, Temperatur = 30 °C, Enthalpie h30 = 125,83 kJ/kg, Entropie s30 = 436,8 J/kg K
Irreversible Mischung
Der thermodynamische Zustand der irreversiblen Mischung (adiabat, keine Abgabe von Arbeit) ergibt sich aus dem Energieerhaltungssatz:
- HM = H10 + H30, hM = (m10*h10 + m30*h30)/(m10 + m30), hM = 83,97 kJ/kg
Mit den Zustandsgrößen Enthalpie und Druck ergeben sich weitere Zustandsgrößen des Mischungszustandes:
- Temperatur tM = 19,99 °C (293,14 K), Entropie sM = 296,3 J/kg K
Reversible Mischung
Bei einer reversiblen Vermischung (dSirr = 0) nimmt die Entropie des Gesamtsystems nicht zu, sie ergibt sich aus der Summe der Entropien der Teilsysteme:
- SM = S10 + S30 + dSirr, sM = 293,9 J/kg K
Mit den Zustandsgrößen Entropie und Druck ergeben sich weitere Zustandsgrößen des Mischungszustandes:
- Temperatur tM = 19,82 °C (292,97 K), Enthalpie hM = 83,26 kJ/kg
Das Gesamtsystem ist in diesem Fall nicht mehr abgeschlossen, sondern tauscht mit der Umgebung Arbeit aus.
Differenzen zwischen irreversibler und reversibler Mischung: Entropie: 2,4 J/kg K, Enthalpie: 0,71 kJ/kg, Temperatur: 0,17 K
Nach der irreversiblen Mischung ist die Entropie des Gesamtsystems um 2,4 J/kg K größer als beim reversiblen Vorgang. Die reversible Mischung könnte durch den Einsatz einer Carnot-Maschine erreicht werden. Dabei würde dem Teilsystem mit der höheren Temperatur eine unendlich kleine Menge Energie entnommen. In Form von Wärme wird diese Energie an der Systemgrenze bei einer unendlich kleinen Temperaturdifferenz an die Carnot-Maschine übertragen. Auf entsprechende Weise wird die Energie dem Teilsystem mit der tieferen Temperatur zugeführt. Die Temperaturen der beiden Teilsysteme würden sich dadurch immer weiter angleichen und der Carnot-Faktor der Maschine von anfänglich 0,066 gegen Null streben. Die Carnot-Maschine würde dabei dem Gesamtsystem die sonst entwertete Enthalpiedifferenz von 0,71 kJ/kg als mechanische Arbeit entnehmen. Im irreversiblen Fall entspricht diese Energie der innerhalb des Systems dissipierten Arbeit. Durch die dissipierte Arbeit wird die produzierte Entropie vom absoluten Nullpunkt auf die Temperatur von 19,99 °C angehoben.
Mischungsentropie
Abbildung 3 zeigt die Mischung einer braunen Farbe in Wasser. Zu Beginn ist die Farbe ungleichmäßig verteilt. Nach längerem Warten nimmt das Wasser eine gleichmäßige Färbung an.
Die Entropie ist ein Maß der Unwissenheit, d. h. der Unkenntnis darüber, in welchem mikroskopischen Zustand sich das betrachtete System befindet. Als Maß der Unordnung muss man auf die Begrifflichkeit achten. So ist im Bildbeispiel (Abbildung 3) die Flüssigkeit im rechten Glas zwar „ordentlicher“ verrührt, aber durch die große Durchmischung von Wasser- und Farbteilchen herrscht dort eine größere Unordnung. Es gibt dort mehr mikroskopisch mögliche Zustände, in denen das Glas sein könnte. Mithin ist dort die Entropie höher als im linken Glas. Von der Farbe wissen wir, dass sie im rechten Glas überall im Wasser verteilt ist. Das linke Bild in Abbildung 3 sagt uns mehr. Wir können Bereiche ausmachen, in denen Farbe in hoher Konzentration anzutreffen ist, oder Bereiche, die frei sind von Farbe.
Josiah Willard Gibbs wies auf den Widerspruch hin, dass der Entropiezuwachs auch auftreten sollte, wenn statt der Tinte Wasser ins Wasserglas gegossen wird (Gibbssches Paradoxon).
Die Zahl der Anordnungen der Farbmoleküle am Anfang ist deutlich geringer als die, wenn sich die Farbe im gesamten Volumen verteilen kann. Denn die Farbmoleküle sind nur auf wenige Bereiche konzentriert. Im rechten Bild von Abbildung 3 können sie sich im gesamten Glas aufhalten. Die Entropie ist hier größer, weshalb das System im Laufe der Zeit dieser Gleichverteilung zustrebt.[29]
Entropiezunahme bei irreversibler und reversibler isothermer Expansion
Die spontane Expansion eines Gases in ein leeres Volumen ist irreversibel, daher ist zur Berechnung der Entropieänderung eine direkte Anwendung der Definition von Clausius nicht möglich. Hierfür wird ein reversibler Ersatzprozess benötigt, der vom gleichen Anfangszustand zum gleichen Endzustand führt. Für einen Prozess bei konstanter Temperatur erhält man die Entropieänderung . Diese Methode beruht darauf, dass die Entropie eine Zustandsgröße ist, ihre Wertänderung also nicht davon abhängen kann, durch welchen Prozess man vom Anfangs- zum Endzustand kommt.

Seien zwei Behälter durch einen Absperrhahn getrennt, siehe Abbildung. Der eine Behälter hat das Volumen und ist mit einem Gas gefüllt, im anderen Behälter herrscht Vakuum. Dann wird der Absperrhahn geöffnet und das Gas verteilt sich gleichmäßig auf das Gesamtvolumen beider Behälter zusammen. Anfangs bilden die Teilchen des Gases den Anfangszustand mit der Entropie . Im Endzustand, wenn nach dem Öffnen sich wieder ein Gleichgewicht eingestellt hat, haben die Teilchen die Entropie . Für ein ideales Gas, so zeigt der Gay-Lussac-Versuch, bleibt dabei die Temperatur konstant, ohne dass Wärme mit der Umgebung ausgetauscht wird. Da die Expansion ohne Gegendruck erfolgt, wird auch keine Arbeit geleistet.




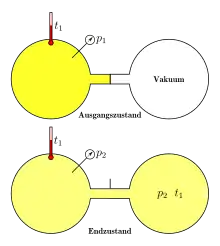
Ein für die Berechnung der Entropieänderung geeigneter Ersatzprozess ist die reversible isotherme Expansion, wie sie z. B. im Carnot-Zyklus eingesetzt wird. Bei diesem Prozess befindet sich das Gas in einem Zylinder mit Kolben und in Kontakt mit einem Wärmebad gleicher Temperatur. Dann drückt das Gas den Kolben nach außen, während ihm gleichzeitig soviel Wärme zugeführt wird, dass die Temperatur gehalten wird. Ist das Gasvolumen von auf angewachsen, wurde bei einem idealen Gas die Arbeit


geleistet, wobei der Druck nach der Zustandsgleichung des idealen Gases eingesetzt wurde. Da mit der Temperatur auch die innere Energie des idealen Gases unverändert bleibt, ist nach dem 1. Hauptsatz die zugeführte Wärme gleich dem negativen der geleisteten Arbeit, also .[30] Daraus folgt für die Entropieänderung:



Wegen ist dies eine Entropiezunahme.

Zum gleichen Ergebnis kommt man auch über die statistische Deutung der Entropie
- .

Darin sind die Phasenraumvolumen für Anfangs- bzw. Endzustand.

Da sich die Temperatur nicht ändert, bleibt das Phasenraumvolumen im Impulsraum konstant und kann herausgekürzt werden. Es bleibt zu berechnen, um welchen Faktor der Ortsanteil des Phasenraumvolumens sich vergrößert. Diesen Faktor kann man einfach verstehen: er ist der Kehrwert der Wahrscheinlichkeit, die Teilchen zufällig im kleineren Volumen zu finden statt im größeren .[31] Bei einem Teilchen ist die Wahrscheinlichkeit, im Endzustand das Teilchen im Volumen zu finden, gleich . Im Falle von zwei Teilchen wäre diese Wahrscheinlichkeit und bei N Teilchen .[32][33] Also gilt für die Wahrscheinlichkeit, spontan den Ausgangszustand wieder anzutreffen




Diese Wahrscheinlichkeit ist für typischerweise von der Größenordnung .


So ergibt auch die Definition aus der statistischen Physik für den Entropieunterschied den gleichen Wert wie oben, nämlich:
- .

Biomembranen
Gibt man Lipide, bei Lebewesen beispielsweise als Bausteine der Biomembranen vorkommend, in Wasser, so bilden sich spontan geschlossene Membranstrukturen, sogenannte Vesikel. Da hier Temperatur und Druck gegeben sind (Wärmebad und Druckensemble), ist das thermodynamische Potential, das ein Minimum anstrebt, die freie Enthalpie . Die Enthalpie lässt sich experimentell kalorimetrisch nachweisen, ist also messbar, und sie ist positiv. Da der Prozess spontan abläuft, muss aber negativ sein; d. h., die Entropie muss steigen. Dies ist auf den ersten Blick verwirrend, da die Entropie meistens die Ursache dafür ist, dass sich Stoffe vermischen (Mischungsentropie). Die Entropiezunahme liegt in einer besonderen Eigenschaft des Wassers begründet. Es bildet zwischen den einzelnen Wassermolekülen Wasserstoffbrückenbindungen aus, die ständig fluktuieren und somit einen hohen Beitrag zur Entropie des Wassers leisten. Um die langen Fettsäureketten der Lipide herum entsteht bei deren Lösung in Wasser ein größerer Bereich, in dem keine Wasserstoffbrückenbindungen mehr gebildet werden können. In den Bereichen um die Fettsäureketten herum fehlt der Entropiebeitrag der Wasserstoffbrücken, so dass die Entropie insgesamt abnimmt. Diese Abnahme ist erheblich größer als die durch das bloße Vermischen des Wassers und des Lipids zu erwartende Zunahme. Wenn sich die Fettsäureketten zusammenlagern, können mehr Wasserstoffbrücken gebildet werden, und die Entropie steigt. Man könnte dies auch so formulieren, dass die Fähigkeit des Wassers, fluktuierende Wasserstoffbrücken zu bilden, die Lipide aus der Lösung treibt. Letztlich ist diese Eigenschaft auch mit die Ursache für die schlechte Löslichkeit vieler unpolarer Stoffe, die die Bildung von Wasserstoffbrückenbindungen stören, und für die Senkung der Oberflächenspannung des Wassers durch Tenside.



Berechnung und Verwendung tabellierter Entropiewerte
Die molare Entropie Smol bei einer bestimmten Temperatur T2 und bei konstantem Druck p erhält man mit Hilfe der molaren Wärmekapazität cp(T) durch Integration vom absoluten Nullpunkt bis zur aktuellen Temperatur:


Dazu kommen noch Entropieanteile bei Phasenübergängen. Nach Planck wird die Entropie ideal kristallisierter, reiner Festkörper am absoluten Nullpunkt gleich null gesetzt (Gemische oder frustrierte Kristalle behalten dagegen eine Restentropie). Unter Standardbedingungen spricht man von der Standardentropie S0. Auch nach der statistischen Betrachtungsweise hängen Entropiewert und Wärmekapazität miteinander zusammen: Eine hohe Wärmekapazität bedeutet, dass ein Molekül viel Energie speichern kann, und das kann z. B. auf einer großen Zahl niedrig liegender und daher leicht erreichbarer Energieniveaus beruhen. Entsprechend viele unterschiedliche Verteilungsmöglichkeiten auf diese Niveaus gibt es dann auch für die Moleküle und das führt auch auf einen hohen Entropiewert für den wahrscheinlichsten Zustand.
In elektrochemischen Reaktionen ergibt sich die Reaktionsentropie ∆S aus der gemessene Änderung von dE (elektromotorische Kraft) mit der Temperatur:
- (z = Ladungszahl, F = Faraday-Konstante)

Die Entropieänderung bei idealen Mischungen erhält man mit Hilfe der Molenbrüche xi der beteiligten Substanzen:

wobei sich in realen Mischungen noch eine Zusatzentropie durch die Veränderung der zwischenmolekularen Kräfte beim Mischen ergibt.
Entstehen bei einer chemischen Reaktion neue Moleküle, dann tritt die höchste Entropie in einem ganz bestimmten Gleichgewichtszustand auf, bei dem sich die Moleküle sowohl auf die Edukt- wie auch auf die Produktniveaus verteilen können. Über die folgende Beziehung, in der die Differenzen der Standard-Entropiewerte ∆S0 der beteiligten Substanzen eine wesentliche Rolle spielen, kann die Gleichgewichtskonstante K berechnet werden:

(das ∆ bedeutet in diesem Fall die Änderung der Größe bei vollständigem Reaktionsablauf). Woran man bei einem spontanen Vorgang (z. B. chemischen Reaktionen, Lösungs- und Mischungsvorgängen, Einstellung von Phasengleichgewichten und deren Temperaturabhängigkeit, Osmose u. a.) die Intensität dieses Vorgangs abschätzen kann, das ist die Zunahme der gesamten Entropie zwischen Anfangs- und Gleichgewichtszustand, die der Reaktanten und die der Umgebung zusammengenommen (→ chemisches Gleichgewicht). Die spontane Zunahme der Entropie wiederum ist eine Folge der ständigen Bewegung der Moleküle.
Kurz: Die Standard-Entropie von Stoffen kann aus dem Verlauf der Wärmekapazität mit der Temperatur berechnet werden. Die Kenntnis tabellierter Entropiewerte ermöglicht (zusammen mit den Reaktionsenthalpien) die Voraussage des chemischen Gleichgewichts.
Quantenmechanik
In der Quantenstatistik ist ein Mikrozustand ein reiner Zustand, der durch einen Vektor im Hilbertraum des Vielteilchensystems gegeben ist. Wie in der klassischen statistischen Mechanik ist dies ein Raum mit außerordentlich vielen Dimensionen, selbst wenn dem einzelnen Teilchen nur wenige verschiedene Energieeigenzustände zur Verfügung stehen. Zum Beispiel gibt es bei der magnetischen Kernresonanz für jeden Protonenspin nur zwei Energieeigenzustände, damit aber einen Hilbertraum mit doppelt so vielen Dimensionen, wie Protonen in der Probe sind (etwa in einem kleinen Wassertröpfchen). Der zugehörige Makrozustand ist ein gemischter Zustand, der durch einen statistischen Operator oder auch Dichteoperator beschrieben wird.



Dieser enthält alle Informationen über das System, die durch eine ideale Messung zugänglich sind (das ist viel weniger als bei dem reinen Zustand , dem Mikrozustand). Der Makrozustand ist klassisch gegeben durch ein Ensemble von denjenigen Mikrozuständen, die mit bestimmte „typische makroskopische Größen“ gemein haben, wie z. B. Energie, Volumen und Teilchenzahl. Die Verteilung der Mikrozustände im Phasenraum ist klassisch durch eine Verteilungsfunktion gegeben. An deren Stelle tritt in der quantenmechanischen Beschreibung der Dichteoperator:
- .

Falls die Zustände alle orthogonal sind, ist die Wahrscheinlichkeit, dass sich das betrachtete System im „reinen“ quantenmechanischen Zustand befindet.

Der Erwartungswert einer Observablen auf dem durch den Dichteoperator beschriebenen Zustandsgemisch ist gegeben durch eine Spurbildung:
- .

Die Spur eines Operators ist folgendermaßen definiert: für eine beliebige (vollständige) Basis .


Von-Neumann-Entropie
Die Von-Neumann Entropie (nach John von Neumann) ist definiert als Erwartungswert des Dichteoperators:
- .

Multipliziert man diese dimensionslose Von-Neumann-Entropie mit der Boltzmann-Konstanten , so erhält man eine Entropie mit der gewöhnlichen Einheit.
Die Entropie ist über die Wahrscheinlichkeiten der einzelnen reinen quantenmechanischen Zustände im Makrozustand gegeben durch
- ,

wobei die Wahrscheinlichkeit ist, im i-ten Mikrozustand zu sein. Die Wahrscheinlichkeiten können Werte zwischen und annehmen. (Die Singularität des Logarithmus im Falle ist unerheblich, weil .) Somit ist und die Entropie positiv semidefinit. Liegt das Gemisch in einem reinen Zustand vor, hat eine dieser Wahrscheinlichkeiten den Wert , während alle anderen den Wert Null annehmen. In diesem Fall ist die Entropie Null, hat also den Minimalwert. Positive Werte der Entropie erhält man, wenn mehr als ein Mikrozustand eine von Null verschiedene Wahrscheinlichkeit haben.






Als Beispiel nehmen wir ein Spinsystem mit vier Elektronen. Spin und magnetisches Moment sind antiparallel. Das heißt, das magnetische Moment eines nach unten zeigenden Spins besitzt im äußeren Magnetfeld die Energie . Die Energie des Systems soll insgesamt sein. Dies führt zu den vier Mikrozuständen:






Daraus folgt, dass die Spinentartung ist mit und wie oben auch hier gilt.



Die obige allgemeine Formel, (*), ist bis auf einen konstanten Faktor identisch mit der Formel für die Shannon'sche Informationsentropie. Das bedeutet, die physikalische Entropie ist auch ein Maß für die Information, die einem durch Kenntnis des Makrozustands zum Mikrozustand fehlt.

Eigenschaften der statistischen Entropie eines quantenmechanischen Zustandes
Seien und Dichteoperatoren auf dem Hilbertraum .



- Invarianz unter unitären Transformationen von (mit )



- Minimum
- Minimum wird bei reinen Zuständen angenommen


- Maximum
- Maximum wird angenommen, wenn alle möglichen Zustandsvektoren mit gleicher Wahrscheinlichkeit auftreten


- mit


- Sei Dichteoperator auf und bzw. reduzierte Dichteoperatoren auf bzw.






Bekenstein-Hawking-Entropie von Schwarzen Löchern
Jacob Bekenstein[34] stellte in seiner Doktorarbeit Ähnlichkeiten zwischen der Physik Schwarzer Löcher und der Thermodynamik heraus. Unter anderem verglich er den Zweiten Hauptsatz der Thermodynamik damit, dass die Oberfläche von Schwarzen Löchern mit einfallender Materie anscheinend immer anwächst und keine Materie entweichen kann. Es ergab sich als Formel für die Entropie
- ,

Hier ist die Oberfläche des Ereignishorizonts, die Gravitationskonstante, die Lichtgeschwindigkeit und die Boltzmannkonstante.



Stephen Hawking kritisierte daran, dass damit das Schwarze Loch auch eine Temperatur besitzen müsse. Ein Körper mit einer nicht verschwindenden Temperatur emittiert jedoch eine Schwarzkörperstrahlung, die der Annahme widerspricht, dass aus dem Schwarzen Loch nichts mehr entweicht. Hawking löste dieses Paradoxon durch die Postulierung der nach ihm benannten Hawking-Strahlung auf: In der quantenmechanischen Beschreibung des Vakuums sind ständig Vakuumfluktuationen aus Teilchen-Antiteilchen-Paaren vorhanden. Wenn bei einer Paarbildung knapp außerhalb des Ereignishorizonts eines der beiden Partnerteilchen vom Schwarzen Loch „eingefangen“ wird, das andere aber entkommt, entspricht dies physikalisch einer thermischen Strahlung des Schwarzen Lochs.[35][36] Zur Realität solcher thermischer Strahlung ist zu sagen, dass Beobachter in unterschiedlichen Bezugssystemen unterschiedliche Beobachtungen machen, also Temperatur oder Eigentemperatur. Erst die Entdeckung von Hawking, dass ein Beobachter, der von einem schwarzen Loch mit Ereignishorizont bei weit entfernt ist, die Schwarzschild-Temperatur


beobachtet, und Untersuchung einer freien Quantenfeldtheorie in Rindler-Raum-Koordinaten, führte zu der Erkenntnis der Hawking-Strahlung als Evaporation des schwarzen Lochs von Teilchen mit niedrigem Drehimpuls, während andere mit höheren Drehimpulsen von den Wänden des Lochs reflektiert werden.[37]
Das Schwarze Loch kann sich auflösen, wenn die Energie der abgestrahlten Hawking-Strahlung (durch die die Masse des Schwarzen Lochs abnimmt) für einen ausreichend langen Zeitraum den Energieinhalt der einfallenden Materie übersteigt.[38]
Siehe auch
Literatur
- Skripte
- Georg Job, Regina Rüffler: Physikalische Chemie. Teil 1: Grundzüge der Stoffdynamik. Eduard-Job-Stiftung für Thermo- und Stoffdynamik, September 2008, abgerufen am 10. Dezember 2014 (darin insbesondere Kapitel 2).
- F. Herrmann: Thermodynamik. (PDF; 12,87 MB) Physik III. Abteilung für Didaktik der Physik, Universität Karlsruhe, September 2003, archiviert vom Original; abgerufen am 6. Juni 2020.
- Lehrbücher und Übersichtsartikel
- Klaus Stierstadt, Günther Fischer: Thermodynamik: Von der Mikrophysik zur Makrophysik (Kap. 5). Springer, Berlin, New York 2010, ISBN 978-3-642-05097-8 (eingeschränkte Vorschau in der Google-Buchsuche).
- R. Frigg, C. Werndl: Entropy – A Guide for the Perplexed (PDF; 301 kB). In: C. Beisbart, S. Hartmann (Hrsg.): Probabilities in Physics. Oxford University Press, Oxford 2010. (Überblick über die verschiedenen Entropiebegriffe und deren Verknüpfungen).
- G. Adam, O. Hittmair: Wärmetheorie. 4. Auflage. Vieweg, Braunschweig 1992, ISBN 3-528-33311-1.
- Richard Becker: Theorie der Wärme. 3., erg. Auflage. Springer, 1985, ISBN 3-540-15383-7.
- Arieh Ben-Naim: Statistical Thermodynamics Based on Information: A Farewell to Entropy. 2008, ISBN 978-981-270-707-9.
- Johan Diedrich Fast: Entropie. Die Bedeutung des Entropiebegriffes und seine Anwendung in Wissenschaft und Technik. 2. Aufl. Hilversum 1960.
- Ulrich Nickel: Lehrbuch der Thermodynamik. Eine anschauliche Einführung. 3., überarbeitete Auflage. PhysChem, Erlangen 2019, ISBN 978-3-937744-07-0.
- E. P. Hassel, T. V. Vasiltsova, T. Strenziok: Einführung in die Technische Thermodynamik. FVTR GmbH, Rostock 2010, ISBN 978-3-941554-02-3.
- Arnold Sommerfeld: Vorlesungen über theoretische Physik – Thermodynamik und Statistik. Nachdruck der 2. Auflage. Harri Deutsch, 1988, ISBN 3-87144-378-6.
- Leonard Susskind and James Lindesay: An Introduction to BLACK HOLES, INFORMATION and the STRING THEORY REVOLUTION, World Scientific, 2005, ISBN 978-981-256-083-4.
- André Thess: Das Entropieprinzip – Thermodynamik für Unzufriedene. Oldenbourg-Wissenschaftsverlag, 2007, ISBN 978-3-486-58428-8.
- Wolfgang Glöckner, Walter Jansen, Hans Joachim Bader (Hrsg.): Handbuch der experimentellen Chemie. Sekundarstufe II. Band 7: Mark Baumann: Chemische Energetik. Aulis Verlag Deubner, Köln 2007, ISBN 978-3-7614-2385-1.
- André Thess: Was ist Entropie? Eine Antwort für Unzufriedene. In: Forschung im Ingenieurwesen. Band 72, Nr. 1, 17. Januar 2008, S. 11–17, doi:10.1007/s10010-007-0063-7.
- Populärwissenschaftliche Darstellungen
- Arieh Ben-Naim: Entropy Demystified – The Second Law Reduced to Plain Common Sense. World Scientific, Expanded Ed., New Jersey 2008, ISBN 978-981-283-225-2. (populärwissenschaftliche, aber exakte Erklärung auf Grundlage der statistischen Physik).
- H. Dieter Zeh: Entropie. Fischer, Stuttgart 2005, ISBN 3-596-16127-4.
- Eric Johnson: Anxiety and the Equation: Understanding Boltzmann's Entropy. The MIT Press, Cambridge, Massachusetts 2018, ISBN 978-0-262-03861-4.
- Jeremy Rifkin, Ted Howard: Entropy: A New World View. Viking Press, New York 1980 (dt.: Entropie: Ein neues Weltbild. Hofmann & Campe, Hamburg 1984).
Weblinks
- Was ist Entropie? aus der Fernseh-Sendereihe alpha-Centauri (ca. 15 Minuten). Erstmals ausgestrahlt am 4. Aug. 2004.
- Tomasz Downarowicz: Entropy. In: Scholarpedia. (englisch, inkl. Literaturangaben)
- Martin Buchholz: Entropie – Von Kühltürmen und der Unumkehrbarkeit der Dinge Siegerbeitrag der Deutschen Meisterschaft im Science Slam 2010 auf Youtube
- Ulf von Rauchhaupt: Zeit, Tod und schmutziges Geschirr Artikel in der Frankfurter Allgemeinen Zeitung über Ludwig Boltzmann und die Entropie
- Thomas Neusius: Entropie und Richtung der Zeit Kursmaterial, beginnend auf Schulniveau
- Owen Maroney: Information Processing and Thermodynamic Entropy. In: Edward N. Zalta (Hrsg.): Stanford Encyclopedia of Philosophy.
- Nico G. van Kampen: Entropie (PDF; 18 kB) kurze, gut verständliche Erläuterung
- W. A. Kreiner: Thermodynamik und Informationstheorie – Deutungen und Bedeutungsunterschiede im Entropiebegriff. doi:10.18725/OPARU-4097 Eine vergleichende Gegenüberstellung.
- Video: Konzept der Entropie nach CLAUSIUS und BOLTZMANN – wie viel Chaos steckt in einem System?. Jakob Günter Lauth (SciFox) 2013, zur Verfügung gestellt von der Technischen Informationsbibliothek (TIB), doi:10.5446/15662.
Einzelnachweise und Anmerkungen
- Temperaturdifferenzen können vorübergehend entstehen oder sich vergrößern, wenn zwischen den beiden Teilsystemen Phasenübergänge oder chemische Reaktionen möglich sind. Vgl. Erhitzung durch Feuer, Kühlung durch Verdunstung. Auf lange Sicht stellt sich aber immer eine einheitliche Temperatur ein.
- Richard Becker: Theorie der Wärme. Springer, Heidelberg 2013, S. 253 (books.google.de [abgerufen am 16. Juni 2015] Nachdruck von 1961).
- Antoine Laurent Lavoisier: Oeuvres de Lavoisier: Traité élémentaire de chimie, opuscules physiques et chimiques, Band 1. Ministre de L’instruction Publique et des Cultes, 1864 (Seite 410, Original der Bayerischen Staatsbibliothek, digitalisiert 8. Dez. 2009 – Registereintrag aus dem Jahr 1789).
- Roger Hahn: Pierre Simon Laplace, 1749–1827: A Determined Scientist. Harvard University Press, 2005, ISBN 0-674-01892-3 (eingeschränkte Vorschau in der Google-Buchsuche).
- Joseph Black: Lectures on the elements of Chemistry. Edinburgh, 1807 (Original von Michigan State University, digitalisiert 16. Okt. 2013 in der Google-Buchsuche – posthume Veröffentlichung von Vorlesungen aus dem Jahr 1760).
- Pierre Kerszberg: Natural philosophy. In: Knud Haakonssen (Hrsg.): The Cambridge History of Eighteenth-Century Philosophy. Band 1. Cambridge University Press, 2006, ISBN 0-521-86743-6 (eingeschränkte Vorschau in der Google-Buchsuche).
- James D. Stein: Cosmic Numbers: The Numbers That Define Our Universe. Basic Books, 2011 (eingeschränkte Vorschau in der Google-Buchsuche).
- Sadi Carnot: Réflexions sur la puissance motrice du feu et sur les machines propres à développer cette puissance. Bachelier, 1824 (Original von Lyon Public Library, digitalisiert 29. Sept. 2014 in der Google-Buchsuche).
- Rudolf Clausius: Über verschiedene, für die Anwendung bequeme Formen der Hauptgleichungen der mechanischen Wärmetheorie. In: Annalen der Physik und Chemie. Band 125, 1865, S. 353–400 (Textarchiv – Internet Archive [abgerufen am 24. April 2019] auch Vortrag vor der Zürcher Naturforschenden Gesellschaft).
- Rudolf Clausius: Über den zweiten Hauptsatz der mechanischen Wärmetheorie. 1867 (Original von Michigan State University, digitalisiert 29. Juni 2007 in der Google-Buchsuche – Vortrag, gehalten in einer allgemeinen Sitzung der 41. Versammlung deutscher Naturforscher und Aerzte zu Frankfurt am Main am 23. September 1867).
- Wilhelm Ostwald: Die Energie. Verlag von Johann Ambrosius Barth, Leipzig 1908, S. 77.
- Hugh Longbourne Callendar: Proceedings of the Royal Society of London. Series A: Containing Papers of a Mathematical and Physical Character. Band 134, Nr. 825, 2. Januar 1932, S. xxv (Snippet in der Google-Buchsuche).
- Gottfried Falk, Wolfgang Ruppel: Energie und Entropie. Springer-Verlag, 1976, ISBN 3-540-07814-2.
- Tomasz Downarowicz: Entropy. In: Scholarpedia. Band 2, Nr. 11, 2007, S. 3901, doi:10.4249/scholarpedia.3901 (revision #126991).
- Roman Frigg and Charlotte Werndl: Entropy – A Guide for the Perplexed. (PDF; 294 kB) Juni 2010, abgerufen am 12. Dezember 2014 (englisch).
- Beim 1. Hauptsatz der Thermodynamik tritt im Gegensatz zum 2. Hauptsatz ein solcher „integrierender Faktor“ nicht auf. Der 1. Hauptsatz besagt, dass die Summe (!) der zugeführten Arbeit, und der zugeführten Wärme immer das vollständige Differential einer Zustandsfunktion ergibt, der sogenannten Inneren Energie, obwohl die zwei einzelnen Differentiale nicht vollständig sind. Bei der Funktion wird im Gegensatz zu auch nicht unterschieden, ob die Wärmezufuhr reversibel oder irreversibel erfolgt.
- Hans Dieter Baehr, Stephan Kabelac: Thermodynamik – Grundlagen und technische Anwendungen. 16. Auflage. Springer Vieweg, Braunschweig 2016, ISBN 978-3-662-49567-4, 3.1.2 Die Formulierung des 2. Hauptsatzes durch Postulate und 3.1.3 Die Entropiebilanzgleichung für geschlossene Systeme, S. 92–101.
- E. T. Jaynes: Gibbs vs. Boltzmann Entropies. In: American Journal of Physics. Band 33 (Ausgabe 5), 1965. S. 398
- H.J.W. Müller-Kirsten, Basics of Statistical Physics, 2nd edition, World Scientific 2013, ISBN 978-981-4449-53-3, pp. 28–30.
- L. Susskind and J. Lindesay: An Introduction to BLACK HOLES, INFORMATION and the STRING THEORY REVOLUTION. World Scientific 2005, ISBN 978-981-256-083-4, S. 69–77.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 54.
- W. A. Kreiner: Entropie – was ist das? Ein Überblick. doi:10.18725/OPARU-2609
- Gustav Jaumann: Geschlossenes System physikalischer und chemischer Differentialgesetze. In: Sitzungsber. Akad. Wiss. Wien, Nat.-Naturwiss. Klasse. IIA, Nr. 120, 1911, S. 385–503.
- Erwin Lohr: Entropieprinzip und abgeschlossenes Gleichungssystem. In: Denkschrift der Akad. Wiss. Wien. Nat.-Naturwiss. Klasse, Nr. 93, 1916, S. 339–421 (phys.huji.ac.il [PDF; abgerufen am 11. Juni 2020]).
- Georg Job: Neudarstellung der Wärmelehre – Die Entropie als Wärme. Akademische Verlagsgesellschaft, Frankfurt am Main (Nachdruck von 1961).
- Friedrich Herrmann: Der Karlsruher Physikkurs. 9. Auflage. Teil 1: Energie Impuls Entropie. Aulis Verlag, 2010, ISBN 978-3-7614-2517-6.
- Hans Fuchs: The Dynamics of Heat. Springer, New York 2010.
- Georg Job und Regina Rüffler: Physikalische Chemie - Eine Einführung nach neuem Konzept. Vieweg + Teubner, Wiesbaden 2011.
- F. Herrmann: KPK – Hochschulskripten, Thermodynamik. (PDF; 20,8 MB) 2015, abgerufen am 8. Juni 2020.
- Dem entspricht die Clausius’sche Gleichung (2), die auch folgendermaßen in Worte gefasst werden kann: „In einem abgeschlossenen Thermodynamischen System nimmt die Entropie solange zu, bis Gleichgewicht erreicht ist“.
- Wolfgang Nolting: Grundkurs Theoretische Physik. 9. Auflage. 4/2 Thermodynamik. Springer-Verlag, Berlin, Heidelberg, New York 2016, ISBN 978-3-662-49032-7, 2.7 Entropie als Zustandsfunktion, S. 58–59, doi:10.1007/978-3-662-49033-4 (isotherme Expansion des idealen Gases).
- Klaus Stierstadt: Thermodynamik für das Bachelorstudium. 2 vollst. überarbeitete Auflage. Springer Verlag, Heidelberg 2018, ISBN 978-3-662-55715-0, 2.4, 4.1, 4.2, 4.3, S. 54,85–96, doi:10.1007/978-3-662-55716-7 (z. B. Formel 2.31 oder 4.10).
- Wolfgang Demtröder: Experimentalphysik 1. 9 Auflage. Springer Verlag, 10.3.7 Die Entropie, S. 326–328.
- Frederik Reif: Berkeley Physik Kurs Band 5. 2 Auflage. Vieweg, 1985, S. 79–80.
- Jacob D. Bekenstein: Black holes and entropy. In: Phys.Rev. D, Nr. 7, 1973, S. 2333–2346 (phys.huji.ac.il [PDF; abgerufen am 9. Dezember 2014]).
- Stephen W. Hawking: Particle Creation by Black Holes. In: Commun. Math. Phys. Band 43, 1975, S. 199–220, doi:10.1007/BF02345020.
- Susskind, Lindesay, An Introduction to Black Holes, Information and the String Theory Revolution: The Holographic Universe. World Scientific, Singapur 2004, S. 39–42.
- Susskind, Lindesay, An Introduction to Black Holes, Information and the String Theory Revolution: The Holographic Universe. World Scientific, Singapur 2004, S. 48–49.
- Stephen Hawking: Eine kurze Geschichte der Zeit. 1. Auflage. Rowohlt Verlag, 1988, ISBN 3-498-02884-7 (eingeschränkte Vorschau in der Google-Buchsuche).