Biotinidasemangel
Biotinidasemangel oder spät einsetzender multipler Carboxylasemangel (engl. biotinidase deficiency oder late-onset multiple carboxylase deficiency) ist eine seltene Stoffwechselstörung, die autosomal rezessiv vererbt wird und die dazu führt, dass der Körper das Vitamin Biotin nicht ausreichend recyceln kann. Der Biotinidasemangel gehört zum Formenkreis des Multiplen Carboxylase-Mangels.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| E53.8 | Mangel an sonstigen näher bezeichneten Vitaminen des Vitamin-B-Komplexes |
| ICD-10 online (WHO-Version 2019) | |
Ursache ist ein Defekt des Enzyms Biotinidase. Die Auswirkungen des Biotinidasemangels reichen von Erscheinungen der Haut und Fehlfunktionen des Immunsystems über schwere Stoffwechselentgleisungen bis hin zu Hirnschäden, Koma und Tod.
Eine sehr wirksame Therapie existiert in Form von lebenslanger Behandlung mit Biotin. Ein Test auf Biotinidasemangel ist in vielen Ländern Teil des Neugeborenenscreenings. Man unterscheidet schweren Biotinidasemangel mit einer Enzym-Restaktivität von weniger als 10 % und partiellen Biotinidasemangel mit einer Enzymaktivität von 10 bis 30 %.[2]
Biotinidasemangel zählt zu den so genannten seltenen Krankheiten (englisch orphan disease).
Verbreitung
Weltweit gemittelt, tritt Biotinidasemangel mit einer Häufigkeit von 1:60.000 auf, wobei schwerer und partieller Mangel ähnlich häufig zu finden sind. Rein rechnerisch trägt also einer von 123 Menschen ein entsprechendes defektes Gen. Die Zahlen variieren jedoch von Land zu Land.[3] Den Screeningberichten der Deutschen Gesellschaft für Neugeborenenscreening für die Jahre 2004 bis 2006 ist zu entnehmen, dass in der Bundesrepublik Deutschland die Häufigkeit eines Biotinidasemangels in der Größenordnung von 1:20.000 bis 1:25.000 liegt.[4]
Ursache und Krankheitsentstehung
Genetik
Das Biotinidase-Gen befindet sich auf Chromosom 3 im Bereich p25. Es sind mehr als 60 Mutationen bekannt, die einen Enzymdefekt verursachen. Schwerer Biotinidasemangel mit unter 10 % Enzymaktivität entsteht, wenn beide Allele ein völlig oder fast völlig defektes Enzym codieren. Zu partiellem Biotinidasemangel mit 10–30 % Enzymaktivität kommt es, wenn die eine Genvariante einen schwerwiegenden Defekt, die andere dagegen nur einen Teildefekt mit beträchtlicher Restaktivität produziert.[5] Daneben finden sich in der Literatur aber auch andere Einteilungen.
Stoffwechsel
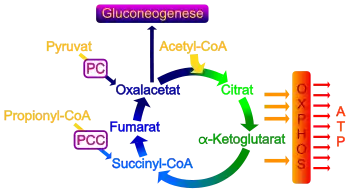
Das Enzym Biotinidase hat die Aufgabe Biocytin, ein Zwischenprodukt des Biotinstoffwechsels, zu spalten und so das darin enthaltene Biotin für den Organismus zurückzugewinnen. Geschieht das ineffizient, geht Biocytin über die Nieren verloren. Durch den überdurchschnittlichen Verlust erschöpfen sich die Biotinvorräte des Körpers trotz ansonsten adäquater Ernährung nach und nach. Dadurch wird die Funktion der vier biotinabhängigen Carboxylasen beeinträchtigt, die grundlegende Aufgaben im Kohlenhydrat-, Protein- und Lipidstoffwechsel erfüllen:[2][6]
- Pyruvat-Carboxylase (Bereitstellung von Oxalacetat für Citratzyklus und Gluconeogenese)
- Propionyl-CoA-Carboxylase (notwendig für die Verstoffwechselung von Aminosäuren und Fettsäuren mit ungradzahligen oder verzweigten Ketten)
- Methylcrotonoyl-CoA-Carboxylase (notwendig für die Verstoffwechselung der Aminosäure Leucin)
- Acetyl-CoA-Carboxylase (notwendig für die Lipogenese)
Infolgedessen wird einerseits der Citratzyklus beeinträchtigt und damit die Energieversorgung der Zellen, andererseits reichern sich in den Mitochondrien Stoffwechselzwischenprodukte in gesundheitsschädlicher Konzentration an, die dann ins Zytosol und schließlich in die Körperflüssigkeiten übertreten. Sind die abnormen Abbauprodukte, oft unter dem Begriff organische Säuren zusammengefasst, im Urin nachweisbar, spricht man von Organoazidurie.
Symptome und Pathologie
Die Symptome ähneln denen eines Biotinmangels. Die Krankheit wirkt sich besonders auf Organe mit hoher Stoffwechselintensität oder Zellteilungsrate aus, wie Gehirn, Muskulatur und Immunsystem. Die Produktion von Hautfett und Prostaglandin[7] kann gestört sein. Es kommt zu Bewegungsstörungen (Ataxie, spastische Parese, Hypotonie), epilepsieartigen Anfällen, Enzephalopathie, Verlust des Gehörs, Schäden am Auge (Gesichtsfeldeinschränkung, Netzhautdegeneration), Schwäche, Appetitlosigkeit, Organoazidurie, Laktatazidose, Ketoazidose, Abnormitäten des Immunsystems, erhöhter Infektanfälligkeit, Konjunktivitis, Dermatitis und Haarausfall (Alopezie).[2][8][9]
Dadurch, dass der Biotinidase-Defekt gleich vier wichtige Enzyme in Mitleidenschaft zieht (drei mitochondriale und ein zytosolisches), sind die Symptome sehr breit gefächert und können sich von Patient zu Patient wesentlich unterscheiden. Keines der genannten Symptome muss bei jedem Patienten auftreten. Auch können verschiedene Gewebetypen verschieden stark vom resultierenden Carboxylasemangel betroffen sein. Die ersten Symptome treten meistens im Alter von drei bis sechs Monaten auf. Die Krankheit kann aber auch früher oder erst wesentlich später beginnen. Die Ausprägung des Krankheitsbildes kann schleichend, plötzlich oder schubweise mit symptomfreien oder symptomärmeren Phasen erfolgen. In einigen Fällen wird metabolischer Stress, beispielsweise durch banale Infekte als Auslöser genannt.[2][10][11]
Neben der in der Literatur häufig beschriebenen Symptomkombination von Krampfanfällen, Hauterscheinungen und, oft erst im fortgeschrittenen Stadium, Organoazidurie und metabolischer Azidose sind auch andere Verlaufsformen möglich. Die folgenden Beispiele zeigen, wie verschieden sich die Erkrankung ausprägen kann:
- Bei Patienten mit partiellem Biotinidasemangel tritt oft nur eine atopische oder eine seborrhoische Dermatitis auf.[2]
- Im Falle von drei Geschwistern starben zwei an fortschreitendem Verfall des Zentralnervensystems und nicht beherrschbaren Infektionen, begleitet von Hautausschlag und Haarausfall. Beim ersten Patienten wurde ein Mangel an Immunglobulin A (IgA) festgestellt, beim zweiten eine erniedrigte Anzahl von T-Lymphozyten. Das dritte Kind litt ebenfalls an mit Candida infizierter Dermatitis, Konjunktivitis, Haarausfall und Schüben von Ataxie, wurde aber nicht immunologisch, sondern metabolisch untersucht. Es zeigte sich ein schubweises Auftreten von Laktatazidose und Organoazidurie.[12]
- Ein Patient zeigte vor allem neurologische Symptome und starb im Alter von 21,5 Monaten. Jedoch fehlten bei ihm Organoazidurie, Haarausfall und Hautausschlag. Post mortem stellte sich heraus, dass die Carboxylase-Aktivität in den Körpergeweben sehr verschieden war mit 42 % in der Niere, 29 % in der Leber, 10 % in den Lymphozyten und 3 % im Gehirn.[13]
- In einem anderen Fall blieb ein Patient bis zum Alter von zehn Jahren unauffällig und symptomfrei. Dann verlor er während eines grippalen Infekts einen Teil seines Sehvermögens. In den folgenden fünf Jahren verstärkten sich die Gesichtsfeldeinschränkungen, neurologische Probleme der Gliedmaßen und körperliche Schwäche kamen hinzu. Es wurden keine Haar- oder Hautprobleme augenfällig, Organoazidurie bestand nur ansatzweise. Nachdem im Alter von 15 Jahren Biotinidasemangel diagnostiziert und Biotin verabreicht wurde, bildeten sich die neurologischen Schäden langsam zurück, das Sehvermögen regenerierte sich zum großen Teil.[11]
Das Krankheitsbild des Biotinidasemangels kann sich in völlig verschiedenen Schweregraden äußern, die nur teilweise von der gemessenen Restaktivität des Enzyms abhängen. Obwohl die Krankheit in vielen Fällen schon früh einen sehr schweren Verlauf nimmt, gibt es auch Erwachsene, bei denen der Biotinidasemangel nur zufällig entdeckt wurde und die nur leichte Auswirkungen des Gendefekts zeigen.[10] Es sind sogar zeitlebens ohne zusätzliche Biotineinnahme asymptomatisch gebliebene Erwachsene mit weniger als 10 % Biotinidase-Aktivität bekannt, auf die man nur deshalb aufmerksam wurde, weil ihre Kinder beim Neugeborenenscreening auffielen. Trotzdem ist auch bei asymptomatisch erscheinenden Individuen die Aktivität der biotinabhängigen Carboxylasen deutlich verringert. Der Zusammenhang zwischen Genotyp und Phänotyp ist noch weitgehend ungeklärt.[5][11][14]
Diagnostik und Differentialdiagnostik
In Abhängigkeit von der Kombination der Symptome beim jeweiligen Patienten ist die Diagnose oft schwierig und langwierig. Da der Biotinidasemangel sich mit sehr verschiedenartigen Erscheinungsformen präsentiert, wobei noch hinzukommt, dass eine vergleichbare Symptomatik auch bei vielen anderen Krankheiten auftritt, sind Labortests unentbehrlich.
Biochemische Indizien
Eine verringerte Aktivität der biotinabhängigen Carboxylasen und das Vorhandensein der so genannten organischen Säuren im Blut, Urin oder Liquor cerebrospinalis sind Sekundär- beziehungsweise Tertiäreffekte, die auf einen Biotinidasemangel hindeuten können. Dabei ist anzumerken, dass eine Organoazidurie nur bei etwa 80 % der Patienten mit Biotinidasemangel auftritt, selbst dann, wenn schon ernste Symptome vorliegen.[2] Andererseits kann ein solcher Befund auch von anderen Defekten des Biotinstoffwechsels oder biotinunabhängigen Organoazidopathien hervorgerufen werden.
Direkte Diagnose
Biotinidasemangel kann durch einen Bluttest nachgewiesen werden, bei dem die Enzymaktivität der Biotinidase direkt bestimmt wird. Auch für das Neugeborenenscreening kommen solche Methoden zum Einsatz.[4] Häufig wird ein colorimetrisches Verfahren mit N-(+)-Biotinyl-4-aminobenzoesäure genutzt.[15] Ein Gentest kann die Diagnose absichern.
Differentialdiagnose
In äußerer Symptomatik und biochemischer Hinsicht dem Biotinidasemangel ähnliche Krankheitsbilder können verursacht werden durch:
- primären Biotinmangel,[1] ernährungsbedingt oder wegen geschädigter Darmflora, Biotinmangel bei Dialyse-Patienten sowie bei Patienten, die langfristig Antikonvulsiva[16] einnehmen.
- andere Formen des Multiplen Carboxylasemangels, wie den Holocarboxylase-Synthetase-Mangel[1] oder Defekte von Biotin-Transportproteinen,[17] zu denen wohl auch die biotin-ansprechende Basalganglienerkrankung gehört. Hier sind zur Behandlung wesentlich höhere Gaben von Biotin erforderlich.
- einen isolierten Mangel der Einzelcarboxylasen. Dort ist eine auf den jeweiligen Enzymdefekt abgestimmte Diät die angezeigte Therapie, Biotin ist nur in Ausnahmefällen wirksam.[18]
- andere Krankheiten der Mitochondrien (Mitochondriopathie, engl. mitochondrial disease).
Behandlung und Heilungsaussicht
Da es sich um einen Gendefekt handelt, ist die Krankheit nicht heilbar im eigentlichen Sinne, es existiert jedoch eine erfolgreiche Behandlungsmethode. Sie besteht in der lebenslangen Gabe von Biotin. Meistens sind 5 bis 20 mg pro Tag ausreichend, es ist aber auch ein Fall beschrieben, bei dem ein Kind 40 mg Biotin täglich benötigte, um symptomfrei zu sein.[19] Durch die hohe Dosis an freiem Biotin umgeht man das defekte Recycling-Enzym. Nebenwirkungen sind nicht bekannt.[2][20]
Wird die Therapie rechtzeitig begonnen und konsequent durchgeführt, ist die Prognose sehr gut. Die Symptome der Haut und des Stoffwechsels bilden sich unter der Behandlung völlig zurück, was auch für viele neurologischen Störungen und Entwicklungsrückstände gilt. Bei längerem Bestehen der Symptome können jedoch irreversible neurologische Schäden wie Beeinträchtigung des Hörvermögens, Sehschäden sowie geistige Behinderung zurückbleiben. Kinder, die über das Neugeborenenscreening diagnostiziert und sofort behandelt werden, entwickeln sich normal.[2][20]
Neugeborenenscreening
In Deutschland ist laut der am 1. April 2005 in Kraft getretenen Richtlinie ein photometrischer Biotinidase-Test fester Bestandteil des Neugeborenenscreenings und eine Leistung der gesetzlichen Krankenkassen. Vorher bestand lediglich eine Empfehlung für den Test, der zu verschiedenen Zeitpunkten in den Bundesländern eingeführt wurde. Die weitere Betreuung positiv getesteter Neugeborener erfolgt am besten in einem spezialisierten Stoffwechselzentrum, entsprechende Kontaktdaten werden von den Screeninglabors bereitgehalten.[21]
Geschichte
Dass manche Stoffwechselerkrankungen, bei denen die Funktion der Carboxylasen beeinträchtigt ist, mit Biotin behandelt werden können, ist seit Anfang der 1970er Jahre bekannt.[22] Bald stellte sich heraus, dass bei den auf Biotin ansprechenden Patienten immer die Funktion mehrerer Carboxylasen gestört ist. Anfang der 1980er Jahre konnte dann als Ursache eines Teiles dieser biotinabhängigen Störungen eine Fehlfunktion des Recycling-Enzyms Biotinidase aufgedeckt werden.[23] Mehr als ein weiteres Jahrzehnt später, ab Mitte der 1990er Jahre, gelang die Bestimmung der Aminosäuren-Sequenz des menschlichen Biotinidase-Enzyms, die Sequenzierung des zugehörigen Gens und die Charakterisierung von ersten Mutationen, deren Zahl bald sprunghaft anstieg.[5] In dieser Zeit verlagerte sich das Hauptinteresse der Forschung auf die Frage, wie aus der Art des Gendefekts auf die Schwere des Krankheitsverlaufs geschlossen werden kann und ob die Biotinidase neben der wohlbekannten Recycling-Funktion eventuell auch eine das Krankheitsbild beeinflussende Transferase-Aktivität besitzt.[24]
Bald nach der Entdeckung des Biotinidase-Defekts als Ursache der damals spät einsetzender multipler Carboxylase-Mangel genannten Krankheit 1982/83 wurde 1984 eine erste für Reihenuntersuchungen taugliche Testmethode der Biotinidase-Aktivität vorgestellt.[15] In den folgenden Jahren liefen in diversen Ländern Pilotprogramme an, die oft zur Folge hatten, dass der Test der Biotinidase-Aktivität in das Programm des Neugeborenenscreenings aufgenommen wurde. Im Jahr 2006 führten schon ungefähr 25 Länder den Biotinidase-Test durch, jedoch nicht immer flächendeckend, wie beispielsweise in den USA, wo 2006 erst in der Hälfte der Bundesstaaten der Test standardmäßiger Bestandteil des Neugeborenenscreenings war.[2]
Einzelnachweise
- E. R. Baumgartner, T. Suormala: Multiple carboxylase deficiency: inherited and acquired disorders of biotin metabolism. In: Int. J. Vitam. Nutr. Res. Band 67, Nr. 5, 1997, S. 377–384 PMID 9350481.
- C. I. Kaye and the Committee on Genetics: Newborn screening fact sheets. In: Pediatrics. Band 118, Nr. 3, September 2006, S. e934–e963, doi:10.1542/peds.2006-1783 (Volltext-PDF, 413 kB).
- B. Wolf: Worldwide survey of neonatal screening for biotinidase deficiency. In: J. Inherit. Metab. Dis. Band 14, Nr. 6, 1991, S. 923–927, PMID 1779651.
- Links zu den Screeningberichten (Memento des Originals vom 12. April 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. der Jahre 2004, 2005 und 2006 bei der Deutschen Gesellschaft für Neugeborenenscreening.
- J. Hymes, C. M. Stanley, B. Wolf: Mutations in BTD causing biotinidase deficiency. In: Hum. Mutat. Band 18, Nr. 5, November 2001, S. 375–381, doi:10.1002/humu.1208.
- J. M. Berg, J. L. Tymoczko, L. Stryer: Biochemie. 6. Auflage. Spektrum Akademischer Verlag, Elsevier, München 2007, ISBN 978-3-8274-1800-5, S. 515f, 551, 697f, 711–713, 746.
- A. Fischer u. a.: Biotin-responsive immunoregulatory dysfunction in multiple carboxylase deficiency. In: J. Clin. Immunol. Band 2, Nr. 1, Januar 1982, S. 35–38, PMID 6212592.
- M. L. Williams: Biotin-responsive multiple carboxylase deficiency and immunodeficiency. In: Curr. Probl. Dermatol. Band 18, 1989, S. 89–92, PMID 2663376.
- H. J. Wastell, K. Bartlett, G. Dale, A. Shein: Biotinidase deficiency: a survey of 10 cases. In: Arch. Dis. Child. Band 63, Nr. 10, Oktober 1988, S. 1244–1249, PMID 3196050 (Volltext).
- J. R. McVoy u. a.: Partial biotinidase deficiency: clinical and biochemical features. In: J. Pediatr. Band 116, Nr. 1, Januar 1990, S. 78–83, PMID 2295967.
- V. T. Ramaekers u. a.: A biotinidase Km variant causing late onset bilateral optic neuropathy. In: Arch. Dis. Child. Band 67, Nr. 1, Januar 1992, S. 115–119, PMID 1739323 (Volltext).
- M. J. Cowan u. a.: Multiple biotin-dependent carboxylase deficiencies associated with defects in T-cell and B-cell immunity. In: The Lancet. Band 2, Nr. 8134, Juli 1979, S. 115–118, PMID 88554.
- E. R. Baumgartner u. a.: Biotinidase deficiency: a cause of subacute necrotizing encephalomyelopathy (Leigh syndrome). Report of a case with lethal outcome. In: Pediatr Res. Band 26, Nr. 3, September 1989, S. 260–266, PMID 2587127.
- T. Baykal u. a.: Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. In: J. Inherit. Metab. Dis. Band 28, Nr. 6, 2005, S. 903–912, PMID 16435182.
- G. S. Heard, J. R. Secor McVoy, B. Wolf: A screening method for biotinidase deficiency in newborns. In: Clin. Chem. Band 30, Nr. 1, Januar 1984, S. 125–127, PMID 6690118.
- D. M. Mock, N. I. Mock, R. P. Nelson, K. A. Lombard: Disturbances in biotin metabolism in children undergoing long-term anticonvulsant therapy. In: J. Pediatr. Gastroenterol. Nutr. Band 26, Nr. 3, März 1998, S. 245–250, PMID 9523856.
- R. Mardach u. a.: Biotin dependency due to a defect in biotin transport. In: J. Clin. Invest. Band 109, Nr. 12, Juni 2002, S. 1617–1623, PMID 12070309 (Volltext).
- M. R. Baumgartner u. a.: Isolated 3-methylcrotonyl-CoA carboxylase deficiency: evidence for an allele-specific dominant negative effect and responsiveness to biotin therapy. In: Am. J. Hum. Genet. Band 75, Nr. 5, November 2004, S 790–800, PMID 15359379 (Volltext).
- P. C. Navarro, A. Guerra, J. G. Alvarez, F. J. Ortiz: Cutaneous and neurologic manifestations of biotinidase deficiency. In: International Journal of Dermatology Band 39, Nr. 5, 2000, S. 363–365, doi:10.1046/j.1365-4362.2000.00841.x.
- P. Weber, S. Scholl, E. R. Baumgartner: Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. In: Dev Med Child Neurol. Band 46, Nr. 7, 2004, S. 481–484, PMID 15230462.
- Derzeit geltende und frühere Richtlinien zum Neugeborenenscreening (Memento des Originals vom 12. April 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. bei der Deutschen Gesellschaft für Neugeborenenscreening.
- D. Gompertz, K. Bartlett, D. Blair, C. M. Stern: Child with a defect in leucine metabolism associated with beta-hydroxyisovaleric aciduria and beta-methylcrotonylglycinuria. In: Arch. Dis. Child. Band 48, Nr. 12, Dezember 1973, S. 975–977, PMID 4765660 (Volltext).
- B. Wolf, R. E. Grier, R. J. Allen, S. I. Goodman, C. L. Kien: Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. In: Clin. Chim. Acta. Band 131, Nr. 3, Juli 1983, S. 273–281, PMID 6883721.
- B. Wolf: Biotinidase: its role in biotinidase deficiency and biotin metabolism. In: J. Nutr. Biochem. Band 16, Nr. 7, Juli 2005, S. 441–445, PMID 15992688.
Weblinks
- Biotinidasemangel. In: Online Mendelian Inheritance in Man. (englisch) (von der Johns-Hopkins-Universität/Baltimore betreute englischsprachige Datenbank mit umfangreichem Literaturüberblick)
- B. Wolf: Biotinidase Deficiency bei GeneReviews (von der Universität von Washington/Seattle betreute englischsprachige Datenbank), letzte Aktualisierung: 25. September 2008