Proteinstruktur
Die Proteinstruktur ist in der Biochemie in verschiedene Strukturebenen gegliedert. Die Einteilung zu einer Hierarchie in Primärstruktur (Aminosäuresequenz), Sekundärstruktur, Tertiärstruktur und Quartärstruktur wurde erstmals 1952 von Kaj Ulrik Linderstrøm-Lang vorgeschlagen.[1] In Bezug auf die räumliche Anordnung eines Proteins wird gleichbedeutend der Begriff Proteinkonformation verwendet.[2] Änderungen der räumlichen Proteinstruktur werden Konformationsänderungen genannt. Dabei ist die Proteinstruktur enorm wichtig für die Funktion des Proteins. Eine fehlerhafte Proteinstruktur kann zum Ausfall der ursprünglichen Proteinfunktion führen.[3]
Die Hierarchie der Strukturebenen
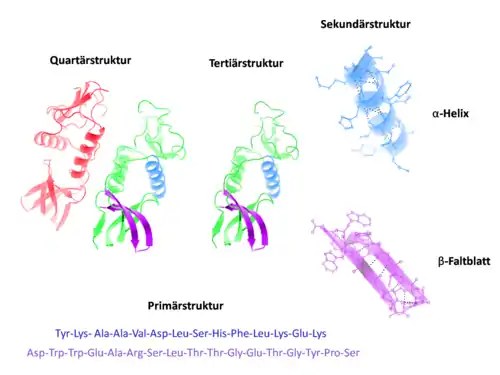
In der Biochemie werden vier hierarchisch angeordnete Strukturebenen in Proteinen unterschieden:
- Primärstruktur – die Aminosäuresequenz (Abfolge der Aminosäuren) der Peptidkette.
- Sekundärstruktur – die räumliche Struktur eines lokalen Bereiches im Protein (z. B. α-Helix, β-Faltblatt).
- Tertiärstruktur – die räumliche Struktur einer Untereinheit.
- Quartärstruktur – die räumliche Struktur des gesamten Proteinkomplexes mit allen Untereinheiten.
Einige Proteine ordnen sich zudem in eine über die Quartärstruktur hinausgehende „Überstruktur“ oder „Suprastruktur“ an. Diese ist molekular genauso praedeterminiert wie die anderen Strukturebenen. Beispiele für Suprastrukturen sind Kollagen in der Kollagenfibrille, Aktin, Myosin und Titin im Sarkomer der Muskelfibrille, sowie Kapsomeren im Kapsid behüllter Viren.
Ausbildung einer Raumstruktur
Der Prozess der dreidimensionalen Raumerfüllung eines Proteins erfolgt teilweise spontan während der Translation, teilweise ist die Mitwirkung von Enzymen oder Chaperonen erforderlich. Auch Liganden beeinflussen die Proteinstruktur, sodass manche Proteine je nach Komplexierung mit Cofaktoren oder Substraten verschiedene Strukturen einnehmen können (siehe: Konformationsänderung). Diese Fähigkeit zur Änderung der Raumstruktur ist für viele Enzymaktivitäten erforderlich.
Störungen in der Bildung einer funktionsfähigen Raumstruktur werden als Proteinfehlfaltungserkrankungen bezeichnet. Ein Beispiel hierfür ist Chorea Huntington. Krankheiten, die auf eine Fehlbildung der Proteinstruktur zurückgehen werden Prionkrankheiten genannt. BSE oder die Alzheimer-Krankheit sind Beispiele für solche Erkrankungen. Auch Diabetes mellitus Typ 2 ist eine Proteinfehlfaltungserkrankung, sie beruht auf einer Fehlfaltung des Amylin.[4] Die räumliche Struktur kann auch durch Denaturierung, aufgrund von Hitze, Säuren oder Basen und ionisierender Strahlung zerstört werden.
Strukturbestimmung
Zur Aufklärung der räumlichen Proteinstruktur stehen verschiedene experimentelle Methoden zur Verfügung:
- Bei der Kristallstrukturanalyse wird – meist mittels Röntgenstrahlen – ein Beugungsbild eines Proteinkristalls erstellt, aus dem sich dann dessen dreidimensionale Struktur errechnen lässt. Die Herstellung der dafür benötigten Einkristalle ist sehr kompliziert und für einige Proteine bisher nicht möglich. Ein weiteres Problem bei diesem Verfahren besteht darin, dass die Struktur der Proteine im Kristall nicht unbedingt der natürlichen Struktur entspricht (crystal packing). Für auswertbare Beugungsbilder ist eine Mindestgröße der Proteinkristalle erforderlich. Um die benötigte Substanzmenge zu erhalten, wird oft auf Proteine zurückgegriffen, die von Bakterien hergestellt wurden. Diese weisen mitunter nicht die bei Proteinen höher Organismen vorhandenen posttranslationalen Modifikationen auf.
- Mittels NMR-Spektroskopie kann die Struktur eines Proteins in Lösung ermittelt werden, was den physiologischen („natürlichen“) Bedingungen des Protein eher entspricht. Da sich Atome des Proteins in diesem Zustand bewegen, ergibt sich keine eindeutige Struktur. Um eine „eindeutige“ Struktur zu erhalten, wird meist über die abgebildeten Strukturen gemittelt. Die NMR-Spektroskopie kann bisher nicht für alle Proteinarten durchgeführt werden. Insbesondere die Größe ist hier ein begrenzender Faktor. Proteine > 30 kDa können bisher nicht analysiert werden, da die NMR-Ergebnisse so komplex sind, dass daraus keine eindeutige Proteinstruktur abgeleitet werden kann.
- Die Struktur ist abhängig von diversen physikochemischen Randbedingungen (wie pH, Temperatur, Salzgehalt, Gegenwart anderer Proteine). Der Stokes-Radius eines nativen Proteins oder eines Proteinkomplexes kann über eine Native-PAGE, eine Größenausschlusschromatographie oder mittels analytischer Ultrazentrifugation ermittelt werden. Diese Methoden können mit einer Quervernetzung oder einem Alanin-Scan kombiniert werden.
Eine umfangreiche Sammlung von Resultaten aus Experimenten zur Strukturbestimmung findet sich in der Protein Data Bank.
Strukturvorhersage
Die Vorhersage räumlicher Proteinstrukturen erzielt gute Ergebnisse, wenn es bereits Proteine mit ähnlicher Sequenz und bekannter Struktur gibt. Dies ermöglicht das sogenannte homology modelling, wobei die neue Sequenz auf die Sequenz, dessen Struktur bekannt ist, abgebildet und damit in die Struktur „eingepasst“ wird. Diese Technik ähnelt dem Sequenzalignment.
Schwieriger ist die Vorhersage, wenn noch keine Strukturen ähnlicher Aminosäuresequenzen bekannt sind. Das Levinthal-Paradox zeigt, dass die Berechnung der energetisch günstigsten Konformation aufgrund der vielen Möglichkeiten nicht durchführbar ist. In der Bioinformatik wurden in den letzten Jahren große Fortschritte gemacht und verschiedene Methoden der de-novo- oder ab-initio-Strukturvorhersage entwickelt. Dennoch existiert bisher keine zuverlässige Methode zur Strukturaufklärung von Proteinen.
Um neue Methoden zur Strukturvorhersage miteinander vergleichen zu können, gibt es seit einigen Jahren den CASP-Wettbewerb (critical assessment of techniques for protein structure prediction). In diesem Wettbewerb werden Aminosäuresequenzen von Strukturen, an denen Kristallographen gerade arbeiten, für die Teilnehmer zur Verfügung gestellt. Die Teilnehmer verwenden ihre eigenen Methoden, um die Strukturen vorherzusagen. Ein Auswertungs-Team vergleicht die Vorhersagen anschließend mit den experimentell ermittelten Strukturen.
Die Strukturvorhersage war bzw. ist auch Ziel mehrerer Projekte des verteilten Rechnens wie z. B. Rosetta@home, POEM@home, Predictor@home und Folding@home sowie des Human Proteome Folding Projects. Das Spiel Foldit macht sich zudem zur Strukturaufklärung die Vorteile des Crowdsourcing zunutze.
Quellen
- Linderstrøm-Lang, K.U. (1952): Proteins and Enzymes. In: Lane Medical Lectures. Bd. 6, S. 1–115. Stanford University Publications, University Series, Medical Sciences, Stanford University Press.
- Christian B. Anfinsen erhielt 1972 den Nobelpreis für Chemie „für seine Arbeiten über Ribonuklease, insbesondere die Verbindung zwischen Aminosäurereihen und biologisch wirksamen Konformationen“ (offizielle Begründung für die Preisvergabe der Königlich Schwedischen Akademie der Wissenschaften)
- Jeremy M. Berg, John L. Tymoczko, Lubert Stryer, Gregory J. Gatto jr.: Stryer Biochemie. 7. Auflage. Springer Spektrum, 2013, ISBN 978-3-8274-2988-9, S. 25–59.
- L. Skora: High-resolution characterization of structural changes involved in prion diseases and dialysis-related amyloidosis. (PDF; 4,3 MB) Dissertation, Georg-August-Universität Göttingen, 2009, S. iii.
Weblinks
- Dr. Ulrich Marsch (TU München): Bioinformatik-Strategie erfolgreich bei der Strukturaufklärung von Membranproteinen. Pressemitteilung. In: idw-online. Informationsdienst Wissenschaft e. V., 28. Oktober 2010, abgerufen am 29. Oktober 2010.