Tuberöse Sklerose
Die tuberöse Sklerose ist eine autosomal-dominante Erbkrankheit, die mit Fehlbildungen und Tumoren des Gehirns, Hautveränderungen und meist gutartigen Tumoren in anderen Organsystemen einhergeht und klinisch häufig durch epileptische Anfälle und kognitive Behinderungen gekennzeichnet ist.
| Klassifikation nach ICD-10 | |
|---|---|
| Q85.1 | Tuberöse (Hirn-)Sklerose
Bourneville-(Pringle)-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
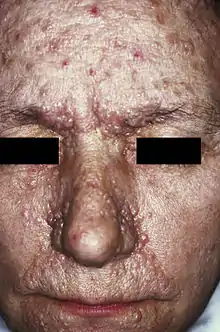
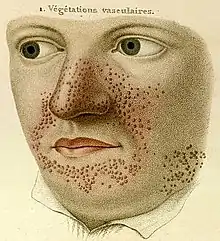
Die Prävalenz der Erkrankung liegt bei Neugeborenen bei etwa 1:8000. Nach den Erstbeschreibern, den französischen Neurologen Désiré-Magloire Bourneville (1840–1909) und Édouard Brissaud (1852–1909)[1] sowie dem britischen Hautarzt John James Pringle (1855–1922) wird diese Erkrankung häufig auch als Bourneville-Pringle-Syndrom oder Bourneville-Brissaud-Pringle-Syndrom bezeichnet. Im englischen Sprachraum hat sich der Begriff Tuberous Sclerosis Complex (TSC) eingebürgert, um den Komplex verschiedener Symptome und Krankheitsbilder bei dieser Erkrankung aus der Gruppe der Phakomatosen hervorzuheben.
Fehlbildungen und Tumoren des Gehirns
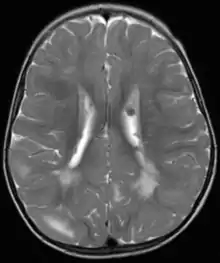
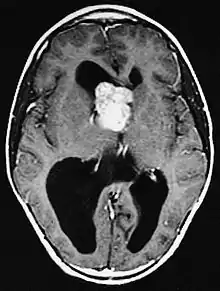
Fehlbildungen und Tumoren des Gehirns werden oft frühzeitig festgestellt. Kortikale glioneuronale Hamartome, die so genannten Tubera (Vorwölbungen) im Bereich der Hirnrinde, gehen häufig mit Epilepsie einher und können kognitive Beeinträchtigungen verursachen, während subependymalen Riesenzellastrozytome und subependymale Knötchen aufgrund ihrer Nähe zum Ventrikelsystem typischerweise zur Entwicklung eines Hydrozephalus führen.
Epilepsie
Epileptische Anfälle sind bei tuberöser Sklerose sehr häufig und können bereits in den ersten Lebensmonaten auftreten. Mit Ausnahme typischer Absencen sind alle Anfallsformen möglich. Die häufigste Anfallsform in der frühen Kindheit sind epileptische Spasmen, häufig wird bei Säuglingen das West-Syndrom diagnostiziert. Insgesamt treten Anfälle bei über 70 % der Kinder mit tuberöser Sklerose auf und können medikamentös nicht immer zufriedenstellend behandelt werden.[2] Ein Zusammenhang zwischen Anfallshäufigkeit und Lernschwierigkeiten konnte nachgewiesen werden.[3] Bei Erwachsenen sind oft rasch sekundär generalisierende fokale Anfälle am häufigsten.
Entwicklungsstörungen
Entwicklungsstörungen mit Beeinträchtigung der Sprach- und Bewegungsentwicklung, aber auch Lernstörungen können Probleme darstellen; mitunter können Verhaltensauffälligkeiten ganz im Vordergrund stehen.[4] Das Ausmaß der Behinderung ist jedoch ausgesprochen heterogen: So wiesen in einer Studie einerseits über die Hälfte der Patienten einen normalen Intelligenzquotienten auf, während bei 31 % der Untersuchten mit einem Intelligenzquotienten unter 21 sehr schwerwiegende Einschränkungen bestanden.[5]
Hautveränderungen
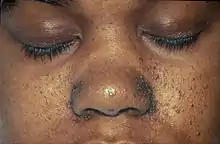
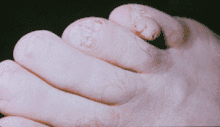
Hautveränderungen kommen in unterschiedlicher Ausprägung vor und treten zum Teil altersabhängig auf. Erste Hautveränderungen sind harmlose Pigmentstörungen, weiße blattförmige Flecken am Körper (so genannte ash-leaf spots) und kommen bei über 80 % der Patienten bereits im ersten Lebensjahr vor. Ihre Erkennung wird durch die Wood-Lampe erleichtert. Im späteren Kindesalter treten typischerweise symmetrisch im Bereich beider Nasolabialfalten gelegene rötliche Papeln hinzu. Hierbei handelt es sich um Angiofibrome, gutartige Hamartome die in Traité théorique et pratique des maladies de la peau (Paris 1826–1827, 1835) von Pierre François Olive Rayer erstmals[6] und von John James Pringle 1890 erstmals als Adenoma sebaceum beschrieben wurden.[7] Typisch sind auch leicht erhabene lederartig verfestigte lumbosakrale Hautläsionen, die als „shagreen patch“ bezeichnet werden (bis zu 40 %). Vom Nagelfalz ausgehende derbe, rötliche fibromatöse Knoten werden als Koenen-Tumor bezeichnet und treten bei 22 % der Patienten in der späten Kindheit auf.[8]
Andere Organsysteme
Außerhalb des Gehirns treten in den Nieren gutartige Tumoren, sogenannte Angiomyolipome, und Nierenzysten auf. Diese Veränderungen machen häufig keine Beschwerden,[9] können aber selten bösartig entarten. Im Herzen werden bei vielen Kindern von Geburt an so genannte Rhabdomyome, Tumore des Muskelgewebes, diagnostiziert. Diese von Friedrich Daniel von Recklinghausen (1833–1910) erstmals beschriebenen Tumoren machen in den meisten Fällen keine ernsthaften Probleme. Sie wachsen bis zur Geburt und bilden sich danach meist zurück; welcher Faktor das bedingt, ist bisher noch nicht bekannt. Bei Beteiligung der Haut treten kutane Angiofibrome (Adenoma sebaceum) auf. Auch die Lunge (Lymphangioleiomyome) und weitere Organe des Körpers können von Tumoren befallen werden.
Vererbung
Die tuberöse Sklerose wird autosomal-dominant vererbt. Das heißt, die Erkrankung kann von einem betroffenen Menschen mit einer Wahrscheinlichkeit von 50 % weiter vererbt werden. Bei 30 % aller betroffenen Menschen erfolgte die Vererbung durch den Vater oder die Mutter. Bei den übrigen 70 % ist die Erkrankung sporadisch aufgetreten und durch eine Neumutation verursacht worden. Auch wenn nur geringe Symptome der tuberösen Sklerose vorliegen, besteht die Möglichkeit, dass Kinder eine stark ausgeprägte Form der tuberösen Sklerose bekommen können. In betroffenen Familien mit Kinderwunsch ist daher eine genetische Beratung durch einen Facharzt für Humangenetik zu empfehlen. Für die Abschätzung der Wiederholungswahrscheinlichkeit kann eine molekulargenetische Untersuchung eines betroffenen Familienmitgliedes eine Hilfe sein, falls die krankheitsverursachende Mutation in einem der Gene für die tuberöse Sklerose (TSC1 auf Chromosom 9 Genlocus q34 und TSC2 auf Chromosom 16 Genlocus p13.3)[10] identifiziert wird und dann gezielt auf diese Mutation hin untersucht werden kann. Mit Hilfe üblicher molekulargenetischer Methoden (Exonsequenzierung und MLPA) sind bei etwa 85 % der Patienten Mutationen oder Deletionen im Bereich des TSC1- oder TSC2-Gens nachweisbar.
Die beiden von den Genen TSC1 und TSC2 codifizierten Proteine gehören zu einem TSC-Proteinkomplex, der ein kritischer negativer Regulator des mTOR-Komplexes 1, das kritisch für die Regelung von Zellwachstum und Zellgröße ist, welches kritische Voraussetzungen für eine Zellteilung sind. Somit wirkt der (intakte) TSC-Proteinkomplex Wachstums- und Mitose-hemmend und somit indirekt als Tumorsuppressoren.[11]
Therapie und Prognose
Eine ursächliche Therapie der tuberösen Sklerose gibt es derzeit nicht, die Behandlung beschränkt sich auf die Symptome, insbesondere auf die der Epilepsie. Zur Therapie der Fehlbildungen und Tumoren des Gehirns stehen neben neurochirurgischen Optionen inzwischen auch immunsuppressive Medikamente mit Hemmung der mTOR-Signalkaskade zur Verfügung.[12] Viele Menschen mit gering ausgeprägter tuberöser Sklerose führen ein weitgehend normales Leben. Bei stärkerer Ausprägung kann die Lebenserwartung insbesondere bei schwerer Epilepsie, ausgeprägten kognitiven Beeinträchtigungen und durch das Auftreten von Tumoren jedoch begrenzt sein.
Literatur
- Kurt Kallenbach (Hrsg.): Kinder mit besonderen Bedürfnissen. Ausgewählte Krankheitsbilder und Behinderungsformen. Ed. Marhold, Berlin 1998, ISBN 3-89166-208-4.
- Immo von Hattingberg: Tuberöse Sklerose (Bourneville). In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 1331.
Weblinks
- Tuberöse Sklerose. In: Online Mendelian Inheritance in Man. (englisch).
- Tuberöse Sklerose Deutschland e. V. mit vielen Informationen
Einzelnachweise
- Bourneville, Brissaud: Encéphalite ou sclérose tubéreuse des circonvolutions cérébrales. In: Archives de neurologie, Paris, 1881, 1, S. 390–412.
- Webb u. a.: Morbidity associated with tuberous sclerosis: a population study. In: Dev Med Child Neurol., 1996, 38(2), S. 146–155. PMID 8603782
- Hunt: Development, behaviour and seizures in 300 cases of tuberous sclerosis. In: J Intellect Disabil Res., 1993, 37, S. 41–51. PMID 7681710.
- Asato, Hardan: Neuropsychiatric problems in tuberous sclerosis complex. In: J Child Neurol. 2004;19(4), S. 241–249. PMID 15163088
- Joinson u. a.: Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. In: Psychol Med. 2003;33(2), S. 335–344. PMID 12622312.
- Barbara I. Tshisuaka: Rayer, Pierre François Olive. In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4, S. 1216 f.
- Pringle: A case of congenital adenoma sebaceum. In: British Journal of Dermatology, 1890, 2, S. 1–14.
- Kane u. a.: Color Atlas & Synopsis of Pediatric Dermatology. McGraw-Hill Professional, 2001, ISBN 0-07-006294-3.
- Webb u. a.: A population study of renal disease in patients with tuberous sclerosis. In: Br J Urol. 1994;74, S. 151–154. PMID 7921930.
- V. Narayanan: Tuberous sclerosis complex: genetics to pathogenesis. In: Pediatr. Neurol. 2003;29(5), S. 404–409. PMID 14684235
- David J. Kwiatkowski, Brendan D. Manning: Molecular Basis of giant cells in tuberous sclerosis complex. In: New England Journal of Medicine, 2014, Band 371, Ausgabe 8 vom 21. August 2014, S. 778–780; doi:10.1056/NEJMcibr1406613.
- DA Krueger, MM Care, K Holland et al.: Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. In: N Engl J Med. Band 363, 2010, S. 1801–1811.