Trametinib
Trametinib ist ein Arzneistoff aus der Gruppe der Kinaseinhibitoren. Es wird allein oder in Kombination mit dem Arzneistoff Dabrafenib zur Behandlung einer bestimmten Hautkrebsart (Melanom) verwendet, die sich in andere Körperregionen ausgebreitet hat oder nicht durch eine Operation entfernt werden kann, und welche eine bestimmte Veränderung (Mutation) an der Position V600 des sogenannten B-Raf-Gens aufweist. Ebenfalls in Kombination mit Dabrafenib wird Trametinib zur Behandlung des fortgeschrittenen nicht-kleinzelligen Lungenkarzinoms (NSCLC) mit B-Raf-V600-Mutation eingesetzt.
| Strukturformel | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
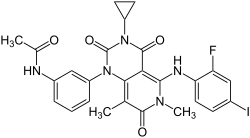 | ||||||||||||||||||||||
| Allgemeines | ||||||||||||||||||||||
| Freiname | Trametinib | |||||||||||||||||||||
| Andere Namen |
| |||||||||||||||||||||
| Summenformel |
| |||||||||||||||||||||
| Externe Identifikatoren/Datenbanken | ||||||||||||||||||||||
| ||||||||||||||||||||||
| Arzneistoffangaben | ||||||||||||||||||||||
| ATC-Code |
L01XE25 | |||||||||||||||||||||
| Wirkstoffklasse |
Kinaseinhibitor | |||||||||||||||||||||
| Wirkmechanismus |
Reversibler, allosterischer Inhibitor der MEK 1 und 2 im MAP-Kinase-Signalweg | |||||||||||||||||||||
| Eigenschaften | ||||||||||||||||||||||
| Molare Masse | 615,40 g·mol−1 | |||||||||||||||||||||
| Schmelzpunkt | ||||||||||||||||||||||
| Löslichkeit |
praktisch unlöslich in Wasser Im pH-Bereich von 2–8[2] | |||||||||||||||||||||
| Sicherheitshinweise | ||||||||||||||||||||||
| ||||||||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | ||||||||||||||||||||||
Patente und Arzneimittelzulassung
Das Erstpatent wurde 2005 von Japan Tobacco Inc.; Tokio, Japan mit der Patentnummer WO 2005121142 (A1)[4] eingereicht. In Deutschland hingegen ist es unter der Nummer DE602005004286 (T2)[5] hinterlegt.
Trametinib wurde im Mai 2013 in den USA unter dem Handelsnamen Mekinist (GlaxoSmithKline) zur Behandlung des nicht-resezierbaren oder metastasierenden Melanom mit B-Raf-V600E- oder V600K-Mutation durch die U. S. Food and Drug Administration (FDA) zugelassen,[6] im Januar 2014 erfolgte die Zulassungserweiterung auf die Kombinationstherapie mit dem B-Raf-Inhibitor Dabrafenib. In den Ländern der EU erfolgte die Zulassung von Mekinist im Juni 2014 und umfasst zusätzlich die Behandlung des fortgeschrittenem nicht-kleinzelligen Lungenkarzinoms.
2015 übertrug GlaxoSmithKline die Zulassung auf Novartis Europharm Ltd.
Chemie
Eigenschaften
Der Arzneistoff Trametinib besitzt keine Stereozentren, verschiedene energiearme Konformationen werden jedoch begünstigt. Die allgemeine räumliche Konfiguration wird durch die folgenden Bilder verdeutlicht.
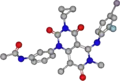 Räumliche Struktur von Trametinib als direkte Aufsicht auf das Grundgerüst der verknüpften Alicyclen
Räumliche Struktur von Trametinib als direkte Aufsicht auf das Grundgerüst der verknüpften Alicyclen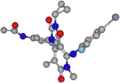 Trametinib aus der Perspektive des halogenierten Aromaten in Richtung des anderen Aromaten durch das Grundgerüst
Trametinib aus der Perspektive des halogenierten Aromaten in Richtung des anderen Aromaten durch das Grundgerüst
Es ist zu erkennen, dass die zwei aromatischen Ringe quasi senkrecht zueinander stehen. Der Cyclopropanring ragt ebenfalls aus der Ebene des Grundgerüsts, das aus zwei verknüpften, aliphatischen Ringen besteht, die leicht gegeneinander verdreht sind.
Synthese
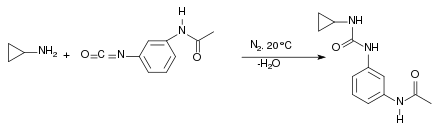
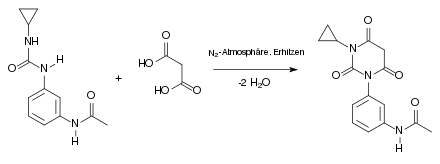
Die Synthese läuft mehrstufig ab.[7] Im ersten Schritt entsteht bei Raumtemperatur in einer Stickstoffatmosphäre aus Cyclopropylamin und dem Isocyanatderivat ein Harnstoffderivat. Dabei greift das Stickstoffatom des Amins nucleophil am Kohlenstoffatom des Isocyanatderivates an:
Beim zweiten Schritt der Synthese handelt es sich um eine Kondensationsreaktion. Man arbeitet in einer Stickstoffatmosphäre und erhitzt dabei. Das Harnstoffderivat reagiert mit der Malonsäure, unter Abspaltung von 2 Molekülen Wasser, zu einem Barbitursäurederivat:
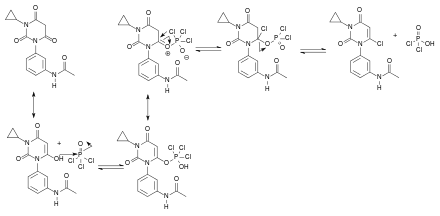
Beim dritten Schritt der Synthese greift das Sauerstoffatom des Barbitursäurederivates nucleophil am Phosphoratom des Phosphoroxychlorids an. Es entsteht ein mesomeriestabilisierter Komplex, bei dem eine Doppelbindung entsteht, an die ein Chloridion des Phosphoroxychlorids angreift. Schlussendlich spaltet sich Dichlorphosphorsäure ab und man erhält das halogenierte Produkt:
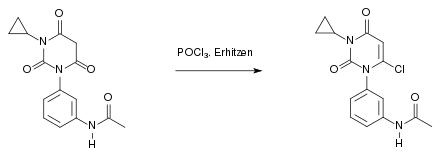
Beim vierten Schritt der Synthese handelt es sich um eine nukleophile Substitution, die nach dem SN1-Mechanismus abläuft. Dabei entsteht als Abspaltungsprodukt Chlorwasserstoff (HCl):
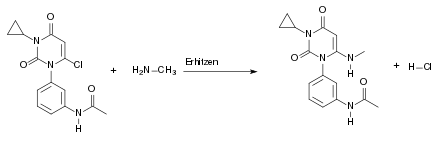
Beim fünften Schritt der Synthese handelt es sich erneut um eine Kondensationsreaktion. Es wird hierbei wieder erhitzt. Dabei reagiert das Produkt aus Teilschritt 4 mit der Methylmalonsäure unter Wasserabspaltung:
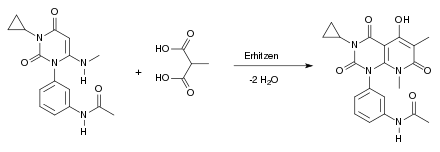
Beim sechsten Schritt der Synthese reagiert Methansulfonylchlorid mit der Hydroxygruppe des Edukts und überführt diese damit in eine gute Abgangsgruppe. Das entstandene HCl wird von der Base Triethylamin abgefangen. Chlorid greift dann mittels einer nukleophilen Substitution (SN1) am Kohlenstoff an. Methylsulfonat wird als Abgangsgruppe abgespalten:
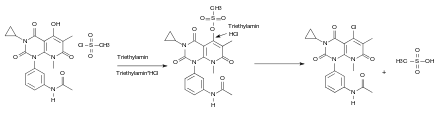

Beim siebten und letzten Schritt der Synthese handelt es sich um eine nukleophile Substitution, die nach dem SN1-Mechanismus abläuft. Dabei entsteht als Abspaltungsprodukt HCl:
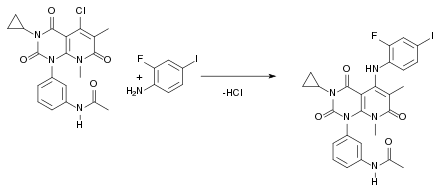
Instrumentelle Methoden
Bisher bekannte, gemessene, Spektren (Stand: Januar 2014) sind ein 1H-NMR Spektrum und ein MS-ESI-Spektrum.
Daten des 1H-NMR Spektrum (DMSO-d6, 400 MHz):
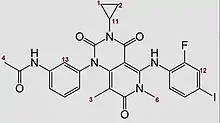
| Chemische Verschiebung δ (ppm) | |
|---|---|
| 1 | 0.63–0.70 (m, 2H) |
| 2 | 0.91–1.00 (m, 2H) |
| 3 | 1.25 (s, 3H) |
| 4 | 2.04 (s, 3H) |
| 5 | 2.58–2.66 (m, 1H) |
| 6 | 3.07 (s, 3H) |
| 7 | 6.92 (t, J=8.8Hz, 1H) |
| 8 | 7.00–7.05 (m, 1H) |
| 9 | 7.36 (t, J=8.2Hz, 1H) |
| 10 | 7.52–7.63 (m, 3H) |
| 11 | 7.79 (dd, J=2,0 , 10.4Hz, 1H) |
| 12 | 10.10 (s, 1H) |
| 13 | 11.08 (s, 1H) |
Daten des MS-ESI-Spektrum: MS/ESI m/e: 616 (M+H), 614 (M-H)
Organische Reaktivitätsanalytik
Zunächst kann mit Hilfe der Beilsteinprobe als allgemeiner Hinweis auf Halogene in organischen Verbindungen das Iod-Atom am halogenierten Aromaten des Trametinib-Moleküls nachgewiesen werden. Das Fluor-Atom bleibt dabei aufgrund der hohen Bindungskraft der C-F-Einfachbindung vermutlich unbeeinträchtigt, was für diese Vorprobe jedoch unerheblich ist.
Nach alkalischer Hydrolyse der Carbonsäureamid-Funktion entsteht Acetat und ein primäres aromatisches Amin als funktionelle Gruppe des Arzneistoffrests.
Nachweismöglichkeiten des primären aromatischen Amins:
- Diazotierung mit salpetriger Säure (in situ mit NaNO2 und HCl) zum aromatischen Diazonium-Salz und anschließender Kopplung mit Bratton-Marshall-Reagenz (bei schwach saurem pH) oder β-Naphthol (im schwach Alkalischen) zum Azofarbstoff.
- Bildung farbiger Imine bzw. „Schiffscher Basen“ durch Reaktion mit Ehrlichs-Reagenz (p-Dimethylaminobenzaldehyd).
- Folin-Reaktion: Nachweis eines farbigen Chinonimins, das bei der Reaktion des primären aromatischen Amins mit Folins-Reagenz (1,2-Naphtochinon-4-sulfonat) entsteht.
- Farbreaktion nach Arylierung mit Sanger-Reagenz (1-Fluor-2,4-dinitrobenzol). Dabei wird das Fluor-Atom des Reagenzes nucleophil durch die primäre Aminofunktion angegriffen und substituiert.
- Organoleptisch, aufgrund des stinkenden Geruchs des Isonitrils, das nach Umsetzung des Amins mit Chloroform im Alkalischen entsteht (Cave, Isonitrile sind kanzerogen!).
Nach erfolgter Hydrolyse von Trametinib und anschließender Aufbereitung des Analysenansatzes besteht die Möglichkeit Essigsäure abzudestillieren und diese bzw. ihre Salze separat mittels folgender Identifikationsreaktionen nachzuweisen:
- Typischer Essigsäure-Geruch; bei Salzen ist dieser erst wahrnehmbar durch sauer reagierende Dämpfe, die nach Protonierung durch z. B. Oxalsäure und Erhitzen des Ansatzes entstehen.
- Nach Veresterung mit Ethanol bei saurem pH tritt ebenfalls ein charakteristischer Geruch auf, nämlich der des Essigsäureethylesters.
- Bildung von widerlich riechendem und giftigen Kakodyloxid, das bei der Reaktion von Salzen der Essigsäure mit Arsenik entsteht.
- Bildung von La(OH)·(CH3COO)2 (= basisches Lanthanacetat) nach Umsetzung von Acetat mit Lanthannitrat. Dabei dient Ammoniak als Lösungsmittel, um den notwendigen alkalischen pH-Bereich von 9-11 einzustellen. Farbgebend bei dieser Reaktion sind wahrscheinlich Iod-Atome, die sich an das gebildete Lanthanacetat adsorbieren können.
- Hydroxamsäure-Reaktion: Im ersten Schritt wird Essigsäure in das Säurechlorid z. B. mit Hilfe von Thionylchlorid überführt. Daran schließt sich die Umsetzung mit Hydroxylamin an, wodurch ein Hydroxamsäure-Derivat entsteht, welches mit Eisen in der Oxidationsstufe drei farbige Komplexe bildet.
Eine Gehaltsbestimmung kann nach der Hydrolysereaktion entweder nitrometrisch (Diazo-Titration), oder bromometrisch (Koppe-Schaar-Titration) erfolgen. Im letzteren Fall werden sechs Äquivalente Brom verbraucht.
Klinische Angaben
Anwendungsgebiete
Trametinib ist angezeigt als Monotherapie oder in Kombination mit Dabrafenib zur Behandlung von erwachsenen Patienten mit nicht-resezierbarem oder metastasiertem Melanom mit einer BRAF-V600-Mutation. Eine Trametinib-Monotherapie hat keine klinische Aktivität bei Patienten gezeigt, deren Erkrankung auf eine vorhergehende Therapie mit einem BRAF-Inhibitor fortschritt.
Trametinib in Kombination mit Dabrafenib ist weiterhin angezeigt zur Behandlung von erwachsenen Patienten mit fortgeschrittenem nicht-kleinzelligen Lungenkarzinom mit einer BRAF-V600-Mutation.[8]
Eingeschlossen in die Zulassungsstudien waren nur Patienten, in denen eine BRAF-V600E- oder V600K-Mutation nachgewiesen worden war. Die Namen der Punktmutationen in den abnormen Genen der Betroffenen rühren daher, dass die Aminosäure Valin (Einbuchstabencode: V) an Position 600 des B-Raf-Proteins gegen Glutaminsäure (E) oder Lysin (K) ausgetauscht sind. Die antitumorale bzw. antiproliferative Wirkung beruht dabei auf der Inhibition der MEK-Kinasen 1 und 2 im MAP-Kinase-Weg.
Art der Anwendung
Mekinist ist oral anzuwenden (Filmtabletten), dabei wird das Dimethylsulfoxid-Solvat des Trametinib (1:1) eingesetzt.
Die Therapie muss abgesetzt werden, falls sich der Krankheitszustand verschlechtert oder unakzeptable unerwünschte Wirkungen auftreten.
Gegenanzeigen und Anwendungsbeschränkungen
Bei einer Überempfindlichkeit gegen Trametinib darf Mekinist nicht verabreicht werden. Nierenkrankheiten haben wahrscheinlich keine klinische Relevanz auf die Pharmakokinetik von Trametinib, da der Arzneistoff hauptsächlich biliär eliminiert wird. Bei milden Formen hepatischer Vorerkrankungen reicht eine Dosisanpassung aus. Studien liegen jedoch bisher nicht vor.
Es wurden bisher keine Studien mit Schwangeren oder stillenden Müttern durchgeführt. Da in Tierstudien reproduktive Toxizität in Form vom totalen Verlust der Schwangerschaft oder Toxizität für den Fötus schon in niedrigeren systemischen Konzentrationen, als für die Therapie notwendig sind, nachgewiesen wurde, sollte auf die Einnahme von Trametinib während der Schwangerschaft verzichtet werden. Es ist nicht bekannt, ob Trametinib an die Muttermilch abgegeben wird. Eine Gefahr für das zu stillende Kind ist nicht ausgeschlossen, weswegen abgewägt werden muss, ob die Therapie mit Trametinib unterbrochen wird oder mit dem Stillen aufgehört wird.
Während und bis zu vier Monate nach der Therapie mit Trametinib ist eine Schwangerschaftsverhütung unabdingbar, um das Risiko einer Schwangerschaft, aus oben genannten Gründen, auszuschließen.
Wechselwirkungen mit anderen Medikamenten
- Auswirkungen von Trametinib auf arzneistoffmetabolisierende Enzyme und Transporter
- In vitro-Studien belegen, dass Trametinib kein Inhibitor folgender Unterfamilien des Cytochrom P450-Enzymsystems ist: CYP1A2, CYP2A6, CYP2B6, CYP2D6 und CYP3A4. Die Hemmung von CYP2C8, CYP2C9 und CYP2C19 gelang erst bei Konzentrationen, die ein Vielfaches der therapeutisch erforderlichen Konzentration darstellen (9- bis zu >100-fache Menge), weswegen keine Interaktionen mit Arzneimitteln, die über diese Enzyme metabolisiert werden, erwartet werden.
- Ebenfalls stellte sich heraus, dass Trametinib in vitro ein Inhibitor der OATP1B1-, OATP1B3-, BCRP-Transporter und P-Glykoprotein ist. Bei der klinisch relevanten, systemischen Konzentration von 0,04 μM ist Trametinib in vivo allerdings kein Inhibitor der zuvor genannten Transportproteine,
- Effekte anderer Arzneistoffe auf Trametinib
- Die Pharmakokinetik von Trametinib wird nicht durch andere Arzneistoffe beeinflusst. Der Metabolismus durch CYP-Enzyme ist vernachlässigbar gering und Trametinib stellt kein Substrat für die Efflux-Transporter P-Glykoprotein oder BCRP dar. Es wird hauptsächlich via hydrolytischer Enzyme, welche allgemein nicht mit Arzneistoffwechselwirkungen in Verbindung gebracht werden, deacetyliert.
- Arzneistoffe, die das PR-Intervall verlängern
- Mekinist kann vermutlich dosisabhängig das PR-Intervall verlängern, was gleichzeitiger Einnahme von Arzneistoffen mit gleicher Wirkung zu berücksichtigen ist. Inbegriffen sind Antiarrythmika, β-Blocker, Calciumkanalblocker (nicht vom Dihydropyridin-Typ), Digitalis-Glykoside, Sphingosin-1-phosphat-Rezeptormodulatoren und einige HIV-Protease-Inhibitoren.
Unerwünschte Wirkungen
Die folgenden Nebenwirkungen haben sich in klinischen Studien bei der Behandlung von 329 Patienten mit oben genannter Indikation, die einmal täglich 2 mg Mekinist oral zu sich nahmen, herausgestellt. Dabei sind in über 99 % aller Fälle mindestens eine der Nebenwirkungen aufgetreten.
- Am häufigsten aufgetretene Nebenwirkungen (≥ 20 %):
- Hautausschlag, akneähnliche Hautentzündung, periphere Ödeme (Lymphödeme), Durchfall, Müdigkeit, Übelkeit, Erbrechen
- Häufige unerwünschte Wirkungen (≥ 1 %):
- Zellulitis, Lungenembolie, Anämie, Atemnot, Lungenentzündung, Erbrechen
- Nebenwirkungen, die zum dauerhaften Therapieabbruch führten (10 %):
- Abfall des Herzzeitvolumens, linksventrikuläre Dysfunktion, Lungenentzündung, Anstieg des Plasmaspiegels der Alanin-Aminotransferase
- Unerwünschte Wirkungen, die zur Dosisreduzierung (26 %) bzw. kurzzeitiger Unterbrechung der Therapie (36 %) führten:
- Hautausschlag, akneähnliche Hautentzündung, periphere Ödeme, Abfall des Herzzeitvolumens, linksventrikuläre Dysfunktion, Durchfall.
Pharmakologische Eigenschaften
Wirkungsmechanismus
Trametinib ist ein reversibler, allosterischer Inhibitor der MEK1 und MEK2 (mitogen-activated extracellular signal-regulated kinases 1 und 2), der sowohl deren Aktivierung, als auch deren Kinaseaktivität hemmt. Die Potenz des kleinen Moleküls wird verdeutlicht durch die IC50-Werte sowohl der unphosphorylierten Formen der Serin-/Threoninkinasen mit 0,7 nM und 0,9 nM, als auch durch die Angaben für den phosphorylierten Zustand mit 13,2 nM und 10,7 nM. MEK-Proteine (=MAPKK) sind upstream-Regulatoren im MAP-Kinase-Weg, der u. a. an der Zellproliferation und -differenzierung sowie Apoptose beteiligt ist. Im Falle des Melanoms mit B-Raf-V600E- oder V600K-Mutation ist dieser intrazelluläre Signalweg, der sowohl das B-Raf-Protein (=MAPKKK), als auch die MEK1 und 2 einschließt, dauerhaft aktiviert, was zum ungezügelten Zellwachstum und unkontrollierter Vermehrung der Zellen führt. Bei Gabe von 1–2 mg Trametinib wurde die Hemmung der phosphorylierten ERK (=MAPK) und Ki-67 (Biomarker für Zellproliferation), sowie ein Anstieg von p27 (Biomarker für Apoptose) beobachtet.[9][10]
Des Weiteren zeigte sich in vitro, dass Trametinib als hochselektiver Inhibitor der MEK1 und MEK2 keine Affinität zu anderen Kinasen bei Konzentrationen von bis zu 10 μM (6154 ng/ml) hat. Weiterhin konnte beim Screening keine signifikante Bindungsaktivität (IC50> 10 μM, 6154 ng/ml) zu zahlreicher Rezeptoren, Enzymen und Ionenkänälen festgestellt werden.
Aufnahme, Verteilung und Ausscheidung
Trametinib wird nach oraler Gabe rasch aufgenommen und erreicht seine maximale Plasmakonzentration nach 1,5 h (tmax). Die Einzelgabe von 2 mg des Arzneistoffs hat eine Bioverfügbarkeit von 72 % und eine Spitzenkonzentration (cmax) im Plasma von 22,2 ng/ml. Das Verteilungsvolumen beträgt 214 Liter. Trametinib ist bis zu 97,4 % an humane Plasmaproteine gebunden.
Trametinib wird in vitro hauptsächlich deacetyliert, aber auch mono-oxygeniert und glucuronidiert. Die Deacetylierung erfolgt wahrscheinlich durch hydrolysierende Carboxylesterasen und Amidasen. Das Cytochrom P450-Enzymsystem ist nicht in den Metabolismus involviert. Die Ausgangssubstanz stellt die im Blut vorwiegend zirkulierende Verbindung dar.
80 % der Dosis werden über die Fäzes, weniger als 20 % über den Urin ausgeschieden. Eine Menge von < 0,1 % der eliminierten Dosis stellt die unmetabolisierte Ausgangsverbindung dar. Die Halbwertszeit beträgt 5,3 Tage und die Plasma-Clearance 3,21 l/h.
Handelsnamen
Monopräparate Mekinist (US)
Weblinks
Literatur
- Keith T. Flaherty, Caroline Robert, Peter Hersey, Paul Nathan, Claus Garbe, Mohammed Milhem, Lev V. Demidov, Jessica C. Hassel, Piotr Rutkowski, Peter Mohr, Reinhard Dummer, Uwe Trefzer, James M.G. Larkin, Jochen Utikal, Brigitte Dreno, Marta Nyakas, Mark R. Middleton, Jürgen C. Becker, Michelle Casey, Laurie J. Sherman, Frank S. Wu, Daniele Ouellet, Anne-Marie Martin, Kiran Patel, Dirk Schadendorf: Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. In: New England Journal of Medicine. Band 367, Nr. 2, 12. Juli 2012, S. 107–114, doi:10.1056/NEJMoa1203421.
- Jeffrey R Infante, Leslie A Fecher, Gerald S Falchook, Sujatha Nallapareddy, Michael S Gordon, Carlos Becerra, Douglas J DeMarini, Donna S Cox, Yanmei Xu, Shannon R Morris, Vijay GR Peddareddigari, Ngocdiep T Le, Lowell Hart, Johanna C Bendell, Gail Eckhardt, Razelle Kurzrock, Keith Flaherty, Howard A Burris, Wells A Messersmith: Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. In: The Lancet Oncology. Band 13, Nr. 8, August 2012, S. 773–781, doi:10.1016/S1470-2045(12)70270-X.
- Gerald S Falchook, Karl D Lewis, Jeffrey R Infante, Michael S Gordon, Nicholas J Vogelzang, Douglas J DeMarini, Peng Sun, Christopher Moy, Stephen A Szabo, Lori T Roadcap, Vijay GR Peddareddigari, Peter F Lebowitz, Ngocdiep T Le, Howard A Burris, Wells A Messersmith, Peter J O Dwyer, Kevin B Kim, Keith Flaherty, Johanna C Bendell, Rene Gonzalez, Razelle Kurzrock, Leslie A Fecher: Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. In: The Lancet Oncology. Band 13, Nr. 8, August 2012, S. 782–789, doi:10.1016/S1470-2045(12)70269-3.
- J. Jing, J. Greshock, J. D. Holbrook, A. Gilmartin, X. Zhang, E. McNeil, T. Conway, C. Moy, S. Laquerre, K. Bachman, R. Wooster, Y. Degenhardt: Comprehensive Predictive Biomarker Analysis for MEK Inhibitor GSK1120212. In: Molecular Cancer Therapeutics. Band 11, Nr. 3, 6. März 2012, S. 720–729, doi:10.1158/1535-7163.MCT-11-0505.
- A. Vultur, J. Villanueva, C. Krepler, G. Rajan, Q. Chen, M. Xiao, L. Li, P A Gimotty, M. Wilson, J. Hayden, F. Keeney, K L Nathanson, M. Herlyn: MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. In: Oncogene. 29. April 2013, doi:10.1038/onc.2013.131.
Einzelnachweise
- Hiroyuki Abe, Shinichi Kikuchi, Kazuhide Hayakawa, Tetsuya Iida, Noboru Nagahashi, Katsuya Maeda, Johei Sakamoto, Noriaki Matsumoto, Tomoya Miura, Koji Matsumura, Noriyoshi Seki, Takashi Inaba, Hisashi Kawasaki, Takayuki Yamaguchi, Reina Kakefuda, Toyomichi Nanayama, Hironori Kurachi, Yoshikazu Hori, Takayuki Yoshida, Junya Kakegawa, Yoshihiro Watanabe, Aidan G. Gilmartin, Mark C. Richter, Katherine G. Moss, Sylvie G. Laquerre: Discovery of a Highly Potent and Selective MEK Inhibitor: GSK1120212 (JTP-74057 DMSO Solvate). In: ACS Medicinal Chemistry Letters. 2, Nr. 4, 14. April 2011, S. 320–324. doi:10.1021/ml200004g.
- MEKINIST (trametinib) tablets, for oral use
- Für diesen Stoff liegt noch keine harmonisierte Einstufung vor. Wiedergegeben ist eine von einer Selbsteinstufung durch Inverkehrbringer abgeleitete Kennzeichnung von N-{3-[3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydropyrido[4,3-d]pyrimidin-1(2H)-yl]phenyl}acetamide im Classification and Labelling Inventory der Europäischen Chemikalienagentur (ECHA), abgerufen am 20. November 2017.
- WO2005121142 (A1) ― 2005-12-22 In: worldwide.espacenet.com.
- DE602005004286 (T2) ― 2009-01-02 In: worldwide.espacenet.com.
- (Seite nicht mehr abrufbar, Suche in Webarchiven)
- document WO2005121142 (A1) ― 2005-12-22 In: worldwide.espacenet.com.
- Mekinist EU Product Information. (PDF) European Medicines Agency, abgerufen am 4. Juli 2017 (englisch).
- A. G. Gilmartin, M. R. Bleam, A. Groy, K. G. Moss, E. A. Minthorn, S. G. Kulkarni, C. M. Rominger, S. Erskine, K. E. Fisher, J. Yang, F. Zappacosta, R. Annan, D. Sutton, S. G. Laquerre: GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. In: Clinical Cancer Research. Band 17, Nr. 5, 1. März 2011, S. 989–1000, doi:10.1158/1078-0432.CCR-10-2200.
- April KS Salama, Kevin B Kim: Trametinib (GSK1120212) in the treatment of melanoma. In: Expert Opinion on Pharmacotherapy. 14, Nr. 5, April 2013, S. 619–627. doi:10.1517/14656566.2013.770475.