Chorea Huntington
Die Chorea Huntington, auch Huntingtonsche Chorea oder Huntington-Krankheit (englisch Huntington’s disease, HD; ältere Namen: Veitstanz, großer Veitstanz, Chorea major) genannt, ist eine unheilbare erbliche Erkrankung des Gehirns, die durch unwillkürliche, unkoordinierte Bewegungen bei gleichzeitig schlaffem Muskeltonus gekennzeichnet ist,[1] in Demenz mündet und zum Tod führt.
| Klassifikation nach ICD-10 | |
|---|---|
| G10 | Chorea Huntington Chorea chronica progressiva hereditaria |
| F02.2* | Demenz bei Chorea Huntington |
| ICD-10 online (WHO-Version 2019) | |
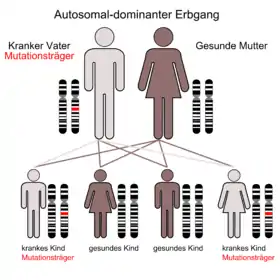
Betroffene leiden an der fortschreitenden Zerstörung eines Bereichs des Gehirns, der für Muskelsteuerung und grundlegende mentale Funktionen wichtig ist, des Striatums. Dort werden Gehirnzellen durch ein fehlerhaftes Eiweiß zerstört, das infolge eines Defekts des Huntingtin-Gens gebildet wird. Die ersten Krankheitserscheinungen treten meistens im 4. Lebensjahrzehnt als Störungen von Körperbewegungen und Gefühlsleben auf. Den weiteren Verlauf prägt ein zunehmender Verlust der Muskelsteuerung einschließlich der Mimik und schließlich der Hirnfunktion insgesamt. Die Erkrankten sterben im Durchschnitt 15 Jahre nach den ersten Symptomen.
Chorea Huntington ist eine autosomal-dominant vererbte neurodegenerative Erkrankung. Mit wenigen Ausnahmen erkranken alle Merkmalsträger. In den 1980er Jahren wurde ein Marker gefunden (James F. Gusella, der unter anderem dafür den König-Faisal-Preis bekam, Nancy Wexler u. a.), der die Entwicklung eines Tests ermöglichte. Seit 1993 lässt sich das Allel, das die Krankheit verursacht, auf dem kurzen Arm des vierten Chromosoms (Genlocus 4p16.3)[2][3][4] nachweisen; dies ist auch beim Ungeborenen durch Amniozentese oder Chorionzottenbiopsie möglich.
Bei der Erforschung der Krankheit spielten zwei auf Privatinitiative gegründete Organisationen eine große Rolle, die von dem Psychologen Milton Wexler nach der Erkrankung seiner Frau 1968 gegründete Hereditary Disease Foundation (HDF), die später von seiner Tochter Nancy Wexler geleitet wurde, und die 1967 von Marjorie Guthrie, der Witwe des an Huntington verstorbenen Woody Guthrie, gegründete Huntington's Disease Society of America (HDSA), aus der auch die Gründung der International Huntingtons Association (IHA) hervorging.[4]
Herkunft des Namens

Die Chorea Huntington (von griechisch χορεία choreia „Tanzen, Tanz“) wurde 1872 von dem New Yorker Arzt George Huntington ausführlich beschrieben. Er beschrieb eine klinische Trias, die lange Zeit Gültigkeit hatte:
- erblich (hereditary nature)
- psychiatrische Auffälligkeiten und Suizidneigung (insanity and suicide)
- schwere Symptome nur im Erwachsenenalter (only in adult life)
Das letzte Kriterium stellte sich später als falsch heraus. Huntington nahm zunächst an, dass die Ausbreitung von Chorea Huntington auf Long Island (Vereinigte Staaten) beschränkt sei. Tatsächlich war die Krankheit aber bereits damals weltweit anzutreffen. Der deutsche Name ist erblicher Veitstanz. Die Bezeichnung Veitstanz (speziell auch Großer Veitstanz[5]), die seit dem 16. Jahrhundert bezeugt ist, hat ihren Ursprung darin, dass als Helfer der heilige Veit (Vitus) angerufen wurde. Wieso gerade dieser Heilige angerufen wurde, ist nicht bekannt. Möglicherweise entstand die Bezeichnung Veitstanz im 15. oder 16. Jahrhundert, als am St.-Veitstag (15. Juni) in Straßburg und anderswo Menschen in großer Zahl von der „Tanzwut“ ergriffen wurden.[6]
Mittlerweile ist in allen ärztlichen und sonstigen Fachkreisen die Bezeichnung Chorea Huntington (im Englischen auch Huntington’s chorea) üblich. Eine der weiteren Bezeichnungen lautet chronische progressive Chorea[7] (lateinisch Chorea chronica progressiva hereditaria).
Epidemiologie
Chorea Huntington ist eine der häufigsten erblich bedingten Hirnstörungen. Eine im Jahr 2012 veröffentlichte Metaanalyse gibt Hinweise auf eine durchschnittliche Prävalenz von 2,71:100.000. Die ausgewerteten Studien ergaben eine Prävalenz von 5,7:100.000 für Europa, Nordamerika und Australien, während sie in Asien bei nur 0,40:100.000 liegt.[8] Die Prävalenz schwankt jedoch von Land zu Land erheblich. So liegt sie in Finnland mit 2,12:100.000 niedriger als im Rest Europas.[9] In Deutschland gibt es offiziell rund 10.000 Betroffene (Stand 2014).[10] In einzelnen Populationen, beispielsweise auf Tasmanien und in der Region Zulia in Venezuela gibt es noch weitaus höhere Prävalenzen, was sich zum Teil auf einzelne aus Europa eingewanderte Personen, die das Gen weitervererbt haben (Gründereffekt), zurückführen lässt. Sehr niedrige Prävalenzen findet man dagegen in Subsahara-Afrika und auch bei Afroamerikanern sowie beispielsweise in Japan, dort liegt die Prävalenz niedriger als 1:100.000. Die Neuerkrankungsrate (Inzidenz) liegt im Mittel bei 4:1.000.000. Männer und Frauen sind gleich häufig betroffen.
Pathophysiologie
Genetik
Chorea Huntington ist eine autosomal-dominant vererbte Krankheit. Dies bedeutet, dass die Nachkommen eines Betroffenen mit einer Wahrscheinlichkeit von etwa 50 % ebenfalls betroffen sind, wenn der phänotypisch erkrankte Elternteil ein mutiertes Allel besitzt (bei zwei mutierten Allelen (Homozygotie) beträgt die Wahrscheinlichkeit nahezu 100 %). Sind beide Elternteile erkrankt und heterozygot, so liegt die Wahrscheinlichkeit für eine Erkrankung der Nachkommen bei etwa 75 %. Generationensprünge kommen nicht vor; Männer und Frauen sind gleich häufig betroffen. Wenn bei der Stammbaumanalyse von Betroffenen kein Blutsverwandter mit Chorea Huntington zu finden ist, wird eine spontan aufgetretene Mutation angenommen.
Bei ungefähr 5 bis 10 Prozent der Patienten liegt eine Neumutation im Gen Huntingtin vor. Das Gen codiert für das gleichnamige Protein und liegt auf dem kurzen Arm von Chromosom 4 (Genlocus 4p16.3). Die Mutation betrifft einen Genbereich, in dessen Sequenz sich das Basentriplett CAG bei der Normalbevölkerung etwa 16 bis 20 mal wiederholt. Durch die zur Chorea Huntington führenden Mutationen wird die Wiederholungshäufigkeit des Tripletts deutlich erhöht, weshalb man auch von einer Trinukleotiderkrankung spricht. Ob sich die Huntingtonsche Krankheit entwickelt, hängt vorrangig von der Anzahl CAG-Wiederholungen (Triplett-Repeats) ab. Bei einer Amplifikation bis etwa drei Dutzend Repeats zeigen sich noch keine Krankheitszeichen. Bei 36 bis etwa 42 CAG-Repeats[11] ist die Penetranz unvollständig, das heißt, nicht alle Menschen dieses Genotyps entwickeln die Krankheit, und auch nach einem Gentest sind keine definitiven Vorhersagen möglich.[12][13] Im Durchschnitt tritt die Erkrankung um so früher in Erscheinung, je häufiger die Wiederholungen sind (Antizipationseffekt). Bei über 60 CAG-Tripletts kann sich eine jugendliche Form der Chorea Huntington manifestieren; ein Ausbruch im vierten Lebensjahr wurde beschrieben. Bei väterlichen (paternalen) Vererbungslinien werden höhere Wiederholungsfrequenzen als bei mütterlicher (maternaler) Vererbung beobachtet (Imprinting-Phänomen).
Molekularbiologie
Ein Triplett CAG der resultierenden mRNA codiert für die Aminosäure Glutamin. Das vom mutierten Huntingtin-Gen exprimierte Protein weist also mehr als die übliche Anzahl aneinandergereihter Glutamin-Reste auf. Möglicherweise handelt es sich dabei um eine „Gain-of-Function-Mutation“, bei der die normale Funktion des Huntingtins erhalten bleibt, das Protein jedoch zusätzlich toxische Eigenschaften hat. Eine hohe Expression von Huntingtin führt zu amyloidähnlichen intrazellulären Ablagerungen (inclusions) von mutiertem Huntingtin, die vom ARF GTPase-aktivierenden Protein1 gefördert werden,[14] wahrscheinlich auch deshalb, weil der Abbau der Proteinmutante durch das Proteasom nicht mehr richtig funktioniert. Andererseits wurde von einigen Arbeitsgruppen auch eine Toxizität des freien mutierten Huntingtins nachgewiesen, sodass die Huntingtin-Aggregate als Schutz angesehen werden können. Die betroffenen Zellen haben einen gestörten Glukosestoffwechsel. Dies führt zu einer gesteigerten Empfindlichkeit gegenüber oxidativem Stress, zudem ist die Sensitivität für den erregenden Neurotransmitter Glutamat erhöht. Diese Zellen besitzen besonders viele Glutamatrezeptoren und haben viele eingehende glutamaterge Verbindungen. Trotzdem ist nur unbefriedigend erklärbar, warum die Toxizität nur in den beschriebenen Arealen nachweisbar ist, obwohl Huntingtin in allen kernhaltigen Zellen gebildet wird.
Die physiologische Funktion von Huntingtin ist nicht vollständig geklärt. Es gibt eine Reihe von Hinweisen dafür, dass es eine wichtige Rolle beim intrazellulären Transport von Vesikeln und Organellen spielt.[15]
Neuroanatomie und Physiologie
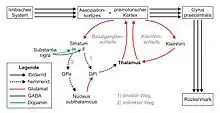
Betroffen ist vor allem das Putamen, welches ein Teil des Corpus striatum in den Basalganglien ist und über einen direkten und über einen indirekten Pfad Einfluss auf den Globus pallidus medialis (interna) nehmen kann.
Der indirekte wirkt dem direkten Pfad entgegen. Der insgesamt hemmende Effekt des indirekten Pfades wird im Gesunden über folgende Stationen erreicht: Die bewegungshemmenden Anteile des Striatums hemmen ihrerseits den Globus pallidus lateralis (externa). Dieser verringert nun seinen hemmenden Effekt auf den Nucleus subthalamicus, wodurch dessen Aktivität verstärkt wird. Da der Nucleus subthalamicus glutamaterge Efferenzen zum Globus pallidus medialis besitzt, fördert er dessen hemmende Wirkung auf den Thalamus.
Bei Menschen mit Chorea Huntington degenerieren in erster Linie GABA/Enkephalin-erge Neurone, d. h., der Anfang des indirekten Pfades ist zerstört. Dies hat zur Folge, dass der Globus pallidus medialis über den direkten Weg schwächer gehemmt wird als bei gesunden Menschen. Da der Globus pallidus medialis seinerseits normalerweise den Thalamus inhibiert, wird dieser nun weniger gehemmt, also aktiviert (= Disinhibition). Die Konsequenz ist eine Übererregung des Thalamus und des Cortex.[16]
Da die indirekten Verbindungen im Verlauf meist zuerst zerstört werden, steht am Anfang der Erkrankung eine Überaktivierung mit überschießenden Bewegungen im Vordergrund. Im weiteren Verlauf gehen auch die direkten Verbindungen verloren, und es dominieren Bewegungsarmut (Akinese) und Steifheit (Rigor).
Krankheitsbild
Verlauf
Erste Symptome der Krankheit zeigen sich meist zwischen dem 30. und 40. Lebensjahr. Das Auftreten von Symptomen ist zwischen dem dritten und dem 75. Lebensjahr beschrieben worden. Patienten mit einem frühen Krankheitsbeginn leiden häufig unter einem schwereren Krankheitsverlauf. Psychische Beschwerden gehen den Bewegungsstörungen oft mehrere Jahre voraus. Die Bewegungsstörungen beginnen meist mit Hyperkinesien (ungewollten Bewegungen) bei verringertem Muskeltonus. Später zeigen sich eher Hypokinesie (Bewegungsarmut) und Erhöhung des Muskeltonus. Eine Verlaufsform, bei der die Bewegungsarmut von Anfang an im Vordergrund steht, wird nach Carl Westphal Westphal-Variante genannt und tritt häufiger bei frühem Krankheitseintritt auf. Die Chorea Huntington schreitet meist mit zunehmender Pflegebedürftigkeit der Betroffenen bis zu 30 Jahre fort. Es kommt durch erschwerte Essensaufnahme (Schluckstörungen) und ständig erhöhten Energieverbrauch häufig zu einer Kachexie (krankhafte Abmagerung). Die meisten Patienten versterben innerhalb von 15 Jahren nach Manifestation der Erkrankung.[17] Das Voranschreiten der Krankheit kann durch Stress beschleunigt werden, umgekehrt haben günstige Lebensumstände mit einer leidensgerechten Aktivierung einen günstigen Einfluss auf den Verlauf der Erkrankung.
Psychische Beschwerden und psychiatrische Symptome
Zu den ersten Erscheinungen der psychischen Veränderung gehören meist Störungen des Affektes und des Antriebes. Diese können auch den Bewegungsstörungen vorangehen. Später können ein unbedachtes und impulsives Verhalten sowie eine Enthemmung in zwischenmenschlichen Beziehungen auftreten. Aufgrund der mangelhaften Kontrolle über die Muskulatur (z. B. des Gesichtes mit Grimassieren) kann der falsche Eindruck eines bereits fortgeschrittenen Persönlichkeitsverlustes entstehen, was bei den Patienten Resignation und Depressionen hervorrufen kann. Besonders in der Frühphase der Erkrankung kann dies zu suizidalem Verhalten führen. Früh treten auch Störungen der visuellen Informationsverarbeitung auf, was z. B. dazu führt, dass die Kranken insbesondere kritische Gesichtsausdrücke ihrer Mitmenschen – wie z. B. Verärgerung – nicht richtig wahrnehmen und so darauf nicht angemessen reagieren können. Im Frühstadium werden leichte Beeinträchtigungen der intellektuellen Fähigkeiten sowie Gedächtnisstörungen oft übersehen. Im Spätstadium der Erkrankung entwickeln die Patienten eine subkortikale Demenz, d. h., es kommt zum Verlust ihrer kognitiven Fähigkeiten.[18] So finden sich Störungen der Merkfähigkeit, damit im Zusammenhang stehend eine Desorientierung und eine Sprachverarmung. Einige Patienten entwickeln Wahnvorstellungen, die dazu führen, dass sie in psychiatrischen Kliniken behandelt werden (psychisch betonter Verlauf).
Bewegungsstörungen
Chorea beginnt meist mit einer zunächst kaum bemerkbaren Bewegungsunruhe der Arme und Beine, des Gesichtes, später des Kopfes sowie des Rumpfes. Diese Unruhe kann sich zu heftigen choreatischen Hyperkinesien steigern. Das sind plötzlich einsetzende, unwillkürliche Bewegungen verschiedener Muskeln, wodurch die Willkürbewegungen unterbrochen werden. Betroffene versuchen zunächst, die choreatischen Bewegungen zu verbergen, indem sie diese in willkürliche Bewegungsabläufe einbauen, z. B. streichen sich nach einer einschießenden Beugebewegung des Armes über das Haar. Zunehmend geraten die Muskelbewegungen aber außer Kontrolle. Beim Vollbild der Erkrankung kommt es zum plötzlichen Grimassieren und zu schleudernden Bewegungen (Chorea) von Armen und Beinen. Sprechen und Schlucken fallen zunehmend schwer (Dysarthrie und Dysphagie). Typischerweise beginnen diese Hyperkinesien in den rumpffernen Teilen der Extremitäten (in den Händen) und im Gesicht, so wird der Mund weit geöffnet, die Zunge weit herausgestreckt und sofort wieder zurückgezogen („Chamäleonzunge“). Im weiteren Verlauf sind auch die rumpfnahen Extremitätenanteile betroffen. Bei Auslösen des Kniesehnenreflexes bleibt das Knie gestreckt (Gordon-Phänomen). Die Bewegungsunruhe verstärkt sich unter seelischer und körperlicher Belastung. Obwohl die unkontrollierten Bewegungen im Schlaf aufhören, nehmen sie bei Ermüdung eher zu. Die anfangs choreatischen Hyperkinesien wandeln sich mit zunehmendem Krankheitsverlauf in Choreoathetose oder Dystonien, wobei durch Erhöhung der Muskelspannung (Muskeltonus) die Gliedmaßen minuten- bis stundenlang in einer manchmal schmerzhaften Fehlstellung verharren. An Stelle des Grimassierens tritt dann eventuell eine Anarthrie auf, d. h., es kann eine völlige Unfähigkeit bestehen, Sprechbewegungen auszuführen, und der Patient ist nicht mehr in der Lage, durch Mimik, Gestik und Sprache zu reagieren. Das Schlucken fällt den Patienten immer schwerer und kann zu lebensbedrohlichen Komplikationen führen, zumal die Patienten durch die Hyperkinesien einen erhöhten Energieverbrauch haben. Dieser kann sich im Endstadium der Erkrankung auf mehr als das Fünffache des normalen Grundumsatzes erhöhen, so dass eine adäquate Versorgung nur noch mit ergänzender parenteraler Nahrungszufuhr möglich ist.
Unabhängig von der Chorea zeigen Menschen mit Chorea Huntington einen Mangel an motorischer Persistenz, also einen unstetigen Muskeltonus. Dieser wird im Englischen auch als milkmaid’s grip umschrieben. Diese Unstetigkeit des Muskeltonus eignet sich besser zur Diagnose des Krankheitsfortschrittes als die Chorea, denn im Gegensatz zur Chorea nimmt sie im Verlauf der Krankheit stetig zu.[13]
Diagnostik
Die Diagnose kann meist klinisch anhand der Symptome gestellt werden. Weitere Möglichkeiten sind eine Kernspintomographie oder Computertomographie. Sie zeigen eine Atrophie des Corpus striatum und hier vor allem des Nucleus caudatus.[19] Diese Atrophie führt zu einer Erweiterung der Seitenventrikel. In der 18FDG-PET zeigt sich eine Störung des Glukosestoffwechsels im Corpus striatum. Eine mögliche Differenzialdiagnose ist CJD (Creutzfeldt-Jakob-Krankheit) sowie vCJD (variant CJD). Darüber hinaus besteht die Möglichkeit, die Diagnose durch genetische Analyse zu sichern. Auch schon bevor sich die ersten Symptome zeigen, kann in einem Institut für Humangenetik eine DNA-Analyse durchgeführt werden, bei ungeborenen Kindern im Rahmen einer Fruchtwasseruntersuchung.[20]
Ethische Probleme der humangenetischen Diagnostik
Es ist heute möglich, lange vor dem Auftreten jeglicher Symptome bei Menschen aus betroffenen Familien eindeutig festzustellen, ob sie den zur Chorea Huntington führenden Gendefekt haben oder nicht und wie viele Repeats der CAG-Sequenz vorhanden sind. Für Kinder eines betroffenen Elternteils, von denen keine DNA-Analyse vorliegt, liegt die Wahrscheinlichkeit des Auftretens der Erkrankung bei 50 %. Sie werden entweder mit Sicherheit oder niemals die Erkrankung bekommen.
Die Entscheidung darüber, ob eine solche Diagnostik gewünscht wird, kann nach einer umfassenden Aufklärung getroffen werden. Eine DNA-Analyse ist vor allem vor einer geplanten Geburt sinnvoll. Mit einer solchen Diagnostik werden auch Informationen über andere Blutsverwandte bekannt. So würde mit einer positiven Diagnostik bei einem Enkel eines Betroffenen auch klar, dass der entsprechende Elternteil betroffen ist, auch wenn dieser noch keine Symptome hat.
Differentialdiagnostik
Abzugrenzen sind Chorea-Huntington-ähnliche Syndrome, z. B.
- Hereditäre neurologische Erkrankungen, z. B. Morbus Wilson, McLeod-Syndrom, Morbus Leigh, Zeroidlipofuszinose
- Autoimmun und paraneoplastisch bedingte choreatische Symptome, z. B. Sydenham Chorea (Chorea minor, Post-Streptokokkeninfektions-Erkrankung), Rasmussen-Syndrom, autoimmun bedingte Enzephalitiden
- Infektiöse Ursachen, z. B. Enzephalopathien bei HIV-Patienten, virale Enzephalitis (Mumps, Masern, Varizella zoster, Herpes simplex), Neuroborreliose, zerebrale Toxoplasmose
- Strukturelle Läsionen der Basalganglien, z. B. bei Schlaganfällen, Neoplasien, abszedierende und demyelinisierende Läsionen
- Metabolische, endokrine und toxische Ursachen, z. B. nicht ketotische Hyperglykämie bei Diabetes mellitus, Elektrolytverschiebungen (Hyper- und Hyponatriämie, Hypokalzämie), Hyperthyreose
- Durch Medikamente und Drogen induzierte Chorea, z. B. bestimmte Antiepileptika, Kalziumkanalblocker, trizyklische Antidepressiva, Antihistaminika
- Andere Ursachen, z. B. Polycythemia vera, essentielle Thrombozythämie[19]
sowie die Chorea-Akanthozytose.
Therapie
Eine Therapie, welche die Krankheit an sich heilt oder dauerhaft aufhält, ist nicht bekannt. Verschiedene Vitamine und Nahrungsergänzungsmittel werden mit unterschiedlichem Erfolg eingesetzt, um die Zellen vor oxidativem Stress zu schützen und so den Krankheitsverlauf zu verlangsamen. Das Medikament Riluzol vermindert die Glutamatausschüttung und soll den Verlauf verlangsamen. Studien weisen darauf hin, dass die pallidale Tiefe Hirnstimulation (THS) ebenfalls positive Effekte insbesondere auf die motorischen Symptome zu haben scheint. Der Krankheitsverlauf kann durch die Stimulation nicht aufgehalten werden, dennoch berichten Betroffene von einer Steigerung der Lebensqualität, da sie weniger auf fremde Hilfe angewiesen sind und weiter aktiv am Sozialleben teilnehmen können.[21][22][23][24][25] Vorklinische Studien lassen auch auf eine Verbesserung weiterer nicht-motorischer Symptome, wie Kognition und Stimmung schließen.[26] Weitere Studien wurden und werden durchgeführt, um die positiven Ergebnisse, die bisher an einzelnen Patienten beobachtet wurden, weiter zu untersuchen.[27]
Sämtliche Therapien werden flankiert von physiotherapeutischer, ergotherapeutischer und logopädischer Behandlung zur Besserung der Bewegungsfähigkeit beziehungsweise der Sprache und Schluckfähigkeit. Gleichzeitig sollten der Patient und auch seine Angehörigen psychiatrisch-psychotherapeutisch behandelt werden. Die Ernährung sollte den erhöhten Energiebedarf, die Schluckbeschwerden und den erhöhten Zuckerbedarf der Patienten berücksichtigen. Einzelne Symptome können ebenfalls je nach Bedarf behandelt werden.
Behandlung der Bewegungsstörungen
Gegen Hyperkinesien werden neben Tetrabenazin auch Dopamin-Antagonisten eingesetzt, meist Tiaprid oder auch Sulpirid. Jedoch zeigen klinische Studien, dass die Einnahme von Tetrabenazin zu einer Verschlimmerung von Depressionen und suizidalen Tendenzen führen könnte.[28] Darüber hinaus verstärkte Tetrabenazin bei manchen Betroffenen die Extrapyramidalen Syndrome.[28][29]
Bei einsetzendem Rigor werden Dopamin-Agonisten oder L-Dopa eingesetzt, können jedoch die Hyperkinesien verstärken. Daher wird die medikamentöse Therapie im Allgemeinen erst eingeleitet, wenn die Bewegungsstörungen den Patienten im Alltag stark beeinträchtigen. Die tiefe Hirnstimulation (THS) hat einen positiven Effekt auf die mit Medikamenten schwer behandelbaren choreatischen Symptome, bei guter Verträglichkeit. Der Einfluss auf Dystonien und anderen Bewegungsstörungen wird in einer klinischen internationalen Studie getestet.[27]
Behandlung psychischer Symptome
Bei psychotischen Symptomen wird auf atypische Neuroleptika zurückgegriffen, gegen depressive Symptome vor allem auf Antidepressiva aus der Gruppe der SSRIs. Bei Schlafstörungen und Angstzuständen können Benzodiazepine zum Einsatz kommen.
Neuroprotektive Behandlung
Es gibt Studien, die eine neuroprotektive Wirkung von Gabapentin auf Patienten mit Chorea Huntington vermuten lassen. Durch dieses Medikament soll die Excitotoxizität des Glutamats auf die Nervenzelle reduziert werden.[30][31]
Pridopidin (früher Huntexil oder ACR16) ist ein Dopamin-Stabilisator, der über Dopamin-Typ-II-Rezeptoren wirkt. In mehreren abgeschlossenen Studien über einen Beobachtungszeitraum von bis zu 26 Wochen konnte Pridopidin keine Wirksamkeit in der Behandlung der Chorea Huntington zeigen.[32] Im Dezember 2020 wurden neue Daten über einen längeren Beobachtungszeitraum veröffentlicht. Patienten, die 52 Wochen mit Pridopidin behandelt wurden, zeigten einen signifikant besseren Verlauf auf der TFC Skala (Total Functional Capacity)[33]. Eine entsprechende Phase-3-Studie ("PROOF-HD") mit mehr als 60 teilnehmenden Zentren wurde initiiert und soll 2021 auch in Italien, Deutschland und Österreich starten.[34][35][36]
Aktuelle Forschung / Huntingtin-Reduzierung
Auch wenn keine ursächliche Therapie bekannt ist, welche die Krankheit heilt oder bremst, wird an der Beeinflussung auslösender Faktoren geforscht. Als „Der Heilige Gral“ gilt zurzeit die sogenannte Huntingtin-Reduzierung.[37] Mit Hilfe verschiedener Ansätze wird versucht, die Produktion dieses Proteins zu reduzieren, und man erhofft sich damit, die Symptome zu vermindern beziehungsweise das Fortschreiten der Krankheit zu bremsen. Während in präklinischen Versuchen dieser Zusammenhang bereits erfolgreich gezeigt werden konnte, ist diese Wirkung beim Menschen nicht bestätigt.
Eine Auswahl (November 2020):
- Tominersen, auch bekannt unter IONIS-HTTRx und RG6042 (Hoffmann-La Roche): Ein Antisense-RNA-Medikament, das regelmäßig intrathekal verabreicht wird. Es soll die Bildung von Huntingtin reduzieren, unterscheidet dabei allerdings nicht zwischen mutiertem und „wildem“ (gesundem) Huntingtin. Phase-1- und Phase-2-Studien wurden erfolgreich abgeschlossen und konnten neben der Sicherheit des Medikaments auch die gewünschte Reduktion des Huntingtin-Proteins zeigen. Ob diese Reduktion auch zu der erhofften Einbremsung der Krankheit führt, wird aktuell in einer weltweit durchgeführten Phase-3-Studie mit ca. 900 Patienten untersucht. (GENERATION HD1[38])
- WVE-120101 / WVE-120102 (Wave Life Sciences) ist wie Tominersen ein Antisense-RNA Medikament, das ebenfalls intrathekal verabreicht wird. Im Gegensatz zu Tominersen wirken diese Substanzen selektiv und sollen nur die mutierten Varianten von Huntingtin reduzieren. Aktuell in Phase 1/2 (PRECISION-HD1[39], PRECISION-HD2[40])
- AMT-130 (uniQure) ist eine Gen-Therapie, die einmalig mit einer Operation am Gehirn durchgeführt wird. Die ersten zwei Patienten wurden im Rahmen einer Phase I/II Studie im Juni 2020 behandelt.[41]
- Weiters werden auch Medikamente in Tablettenform erforscht, wie zum Beispiel PTC518 von PTC Therapeutics. Aktuell wird die Sicherheit des Wirkstoffs in einer Phase-1-Studie überprüft.[42]
- Branaplam ist ein experimentelles Medikament in Tablettenform, das von Novartis eigentlich zur Behandlung von Muskelatrophie der Wirbelsäule entwickelt wird. Bei laufenden Phase-2-Studien soll als Nebeneffekt die Reduktion von mutiertem Huntingtin beobachtet worden sein.[43]
Siehe auch
Weiterführende Literatur
- R. A. Roos: Huntington’s disease: a clinical review. In: Orphanet Journal of Rare Diseases. Band 5, Nummer 1, 2010, S. 40. doi:10.1186/1750-1172-5-40. PMID 21171977. PMC 3022767 (freier Volltext). (Review). (Open Access)
- I. Shoulson, A. B. Young: Milestones in huntington disease. In: Movement disorders. Band 26, Nummer 6, Mai 2011, S. 1127–1133. doi:10.1002/mds.23685. PMID 21626556. (Review).
- A. L. Southwell, P. H. Patterson: Gene therapy in mouse models of huntington disease. In: The Neuroscientist. Band 17, Nummer 2, April 2011, S. 153–162. doi:10.1177/1073858410386236. PMID 21489966. PMC 3131092 (freier Volltext). (Review).
- D. Eidelberg, D. J. Surmeier: Brain networks in Huntington disease. In: The Journal of clinical investigation. Band 121, Nummer 2, Februar 2011, S. 484–492. doi:10.1172/JCI45646. PMID 21285521. PMC 3026742 (freier Volltext). (Review).
- J. Rutishauser: Morbus Huntington: disrupt the fatal attraction. (PDF; 163 kB). In: Schweiz Med Forum. 24, 2002, S. 586–587.
- E. Cattaneo u. a: Das Rätsel der Chorea Huntington. In: Spektrum der Wissenschaft. 2004, S. 60–66.
- Nikola Biller-Andorno: Veitstanz, Chorea major (Neuzeit). In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4, S. 1438 f.
Weblinks
- Leitlinie Chorea der Deutschen Gesellschaft für Neurologie. In: AWMF online (Stand 10/2005)
- Linkkatalog zum Thema Chorea Huntington bei curlie.org (ehemals DMOZ)
- Genetics Home Reference: Huntingtondisease
- Selbsthilfegruppe Deutsche Huntington-Hilfe e. V.
Einzelnachweise
- Nikola Biller-Andorno: Veitstanz, Chorea major (Neuzeit). In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4, S. 1438 f.; hier: S. 1438.
- Darstellung des Gens im NCBI Map Viewer
- Huntington Disease Collaborative Research Group: A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes, Cell, Band 72, 1993, S. 971–983.
- Gillian P. Bates, The molecular genetics of Huntington disease. A History, Nature Reviews Genetics, Band 6, Oktober 2005, S. 766-773
- E. C. Wicke: Versuch einer Monographie des großen Veitstanzes und der unwillkürlichen Muskelbewegung, nebst Bemerkungen über den Taranteltanz und die Berberi. Leipzig 1844.
- Nikola Biller-Andorno: Veitstanz, Chorea major (Neuzeit). In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4, S. 1438 f.; hier: S. 1438.
- Immo von Hattingberg: Systematische Atrophien und Heredodegenerationen des Nervensystems. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 1341–1350, hier: S. 1346.
- T. Pringheim, K. Wiltshire, L. Day, J. Dykeman, T. Steeves, N. Jette: The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. In: Mov. Disord. Band 27, Nr. 9, 2012, S. 1083–1091. PMID 22692795
- J. O.Sipilä, M. Hietala, A. Siitonen, M. Päivärinta, K. Majamaa: Epidemiology of Huntington’s disease in Finland. In: Parkinsonism Relat. Disord. Band 21, Nr. 1, 2015, S. 6–49. PMID 25466405
- Veitstanz soll ins öffentliche Bewusstsein. In: Münchner Merkur. 20. Mai 2014.
- Eintrag zu Chorea Huntington im Flexikon, einem Wiki der Firma DocCheck, abgerufen am 7. Juli 2019.
- E. B. Clabough: Huntington’s disease: the past, present, and future search for disease modifiers. In: Yale J. Biol. Med. Band 86, Nr. 2, 2013, S. 217–233. PMID 23766742
- F. O. Walker: Huntington’s disease. In: The Lancet. Band 369, Nummer 9557, Januar 2007, S. 218–228. doi:10.1016/S0140-6736(07)60111-1. PMID 17240289. (Review).
- Ein Protein fördert krankhafte Ablagerungen bei Chorea Huntington Protein-Interaktionsnetzwerk führte auf die Spur. Pressemitteilung des Max-Delbrück-Centrums für Molekulare Medizin in der Helmholtz Gemeinschaft, 24. September 2004, abgerufen am 8. Oktober 2015.
- J. P. Caviston, E. L. Holzbaur: Huntingtin as an essential integrator of intracellular vesicular trafficking. In: Trends in cell biology. Band 19, Nummer 4, April 2009, S. 147–155. doi:10.1016/j.tcb.2009.01.005. PMID 19269181. PMC 2930405 (freier Volltext). (Review).
- Mahlon R. DeLong: The Basal Ganglia. In: Eric R. Kandel, James H. Schwartz, Thomas M. Jessell: Principles of Neural Science. 2000, ISBN 0-8385-7701-6, S. 860.
- Chorea Huntington. DocCheck.
- K. K. Zakzanis: The subcortical dementia of Huntington’s disease. In: Journal of clinical and experimental neuropsychology. Band 20, Nummer 4, August 1998, S. 565–578. doi:10.1076/jcen.20.4.565.1468. PMID 9892059.
- Gelbe Liste Online: Chorea Huntington - Symptome, Diagnostik, Therapie | Gelbe Liste. Abgerufen am 10. Januar 2022.
- Huntington-Krankheit. Abgerufen am 10. Januar 2022.
- E. Moro, A. E. Lang, A. P. Strafella u. a.: Bilateral globus pallidus stimulation for Huntington’s disease. In: Ann Neurol. Band 56, Nr. 2, August 2004, S. 290–294. doi:10.1002/ana.20183. PMID 15293283.
- B. Biolsi, L. Cif, H. E. Fertit, S. G. Robles, P. Coubes: Long-term follow-up of Huntington disease treated by bilateral deep brain stimulation of the internal globus pallidus. In: J Neurosurg. Band 109, Nr. 1, Juli 2008, S. 130–132. doi:10.3171/JNS/2008/109/7/0130. PMID 18590443.
- M. O. Hebb, R. Garcia, P. Gaudet, I. M. Mendez: Bilateral stimulation of the globus pallidus internus to treat choreathetosis in Huntington’s disease: technical case report. In: Neurosurgery. Band 58, Nr. 2, Februar 2006, S. E383; discussion E383. doi:10.1227/01.NEU.0000195068.19801.18. PMID 16462466
- L. Wojtecki, S. Groiss, S. Ferrea, S. Elben, C. J. Hartmann, S. Dunnett, A. Rosser, C. Saft, M. Südmeyer, C. Ohmann, A. Schnitzler, J. Vesper: A prospective pilot trial for pallidal deep brain stimulation in Huntington’s disease. In: Front. Neurol. 6, 2015, S. 177. doi:10.3389/fneur.2015.00177
- L. Wojtecki, S. J. Groiss, C. J. Hartmann, S. Elben, S. Omlor, A. Schnitzler, J. Vesper: Deep Brain Stimulation in Huntington’s Disease-Preliminary Evidence on Pathophysiology, Efficacy and Safety. In: Brain Sci. 6(3).pii <Teil 2? -->, 30. Aug 2016, S. E38, doi:10.3390/brainsci6030038.
- Y. Temel, C. Cao, R. Vlamings u. a.: Motor and cognitive improvement by deep brain stimulation in a transgenic rat model of Huntington’s disease. In: Neurosci Lett. 406(1-2), 2. Okt 2006, S. 138–141. doi:10.1016/j.neulet.2006.07.036. PMID 16905252.
- Klinische Studie (Phase II): Deep Brain Stimulation (DBS) of the Globus Pallidus (GP) in Huntington’s Disease (HD) (HD-DBS) bei Clinicaltrials.gov der NIH
- Kara J. Wyant, Andrew J. Ridder, Praveen Dayalu: Huntington’s Disease—Update on Treatments. In: Current Neurology and Neuroscience Reports. 17, 2017, doi:10.1007/s11910-017-0739-9.
- M. de Tommaso: Management of Huntington’s disease: role of tetrabenazine. In: Therapeutics and Clinical Risk Management. 7, 2011. S. 123–129, doi:10.2147/TCRM.S17152.
- D. Bonekamp: 1H-Magnetresonanzspektroskopie bei Chorea Huntington unter neuroprotektiver Therapie mit Gabapentin. Dissertation, Medizinische Fakultät Charité – Universitätsmedizin Berlin, 2004.
- C. Cosentino, L. Torres, J. M. Cuba: Gabapentin for Huntington’s disease. In: J Neurology. Band 243, 1996, S: S75-S76.
- ClinicalTrials.gov A Phase 2, to Evaluating the Safety and Efficacy of Pridopidine Versus Placebo for Symptomatic Treatment in Patients With Huntington's Disease
- https://content.iospress.com Effects of Pridopidine on Functional Capacity in Early-Stage Participants from the PRIDE-HD Study
- ClinicalTrials.gov PRidopidine's Outcome On Function in Huntington Disease, PROOF- HD
- https://www.sciadnewswire.com [ https://www.sciadnewswire.com/news/598/huntington-study-group-announces-partnership-prilenia-therapeutics-conduct-global-phase-3 Huntington Study Group Announces a Partnership with Prilenia Therapeutics to Conduct a Global Phase 3 Clinical Study of Pridopidine in Huntington’s Disease]
- YouTube PROOF-HD: Pivotal Huntington Disease Trial Underway
- V. Sung Before the Diagnosis: Symptom hunting Vimeo.com (recorded talk)
- ClinicalTrials.gov A Study to Evaluate the Efficacy and Safety of Intrathecally Administered RO7234292 (RG6042) in Patients With Manifest Huntington’s Disease.
- ClinicalTrials.gov Safety and Tolerability of WVE-120101 in Patients With Huntington's Disease (PRECISION-HD1)
- ClinicalTrials.gov Safety and Tolerability of WVE-120102 in Patients With Huntington's Disease (PRECISION-HD2)
- ClinicalTrials.gov Safety and Proof-of-Concept (POC) Study With AMT-130 in Adults With Early Manifest Huntington Disease
- pipelinereview.com PTC Therapeutics Announces that PTC518 Has Entered into a Phase 1 Clinical Trial for the Huntington's Disease Program
- novartis.com Novartis receives US Food and Drug Administration (FDA) Orphan Drug Designation for branaplam (LMI070) in Huntington’s disease (HD)