Prion
Prionen sind Proteine, die im tierischen Organismus sowohl in physiologischen (normalen) als auch in pathogenen (gesundheitsschädigenden) Konformationen (Strukturen) vorliegen können. Sie vermehren sich nicht durch Teilung, sondern durch induzierte Veränderung benachbarter Moleküle.
Die englische Bezeichnung prion wurde 1982 von Stanley Prusiner vorgeschlagen,[1] der für die Entdeckung der Prionen 1997 den Nobelpreis erhielt. Sie ist ein Kofferwort, abgeleitet von den Wörtern protein und infection[2] und bezieht sich auf die Fähigkeit von Prionen, ihre Konformation auf andere Prionen zu übertragen. Es handelt sich also nicht um Lebewesen, sondern um organische Toxine (Gifte) mit infektiösen Eigenschaften.
Körpereigene Prionen kommen vermehrt im Hirngewebe vor, so dass pathologische Veränderungen schwerwiegende Folgen für den Organismus haben können. Die pathogenen Prionen sind mit großer Wahrscheinlichkeit für die Creutzfeldt-Jakob-Krankheit beim Menschen, BSE („Rinderwahn“) beim Rind oder Scrapie (Traberkrankheit) bei Schafen verantwortlich. Sie gelangen am wahrscheinlichsten durch kontaminierte Nahrung in den Körper (z. B. bei BSE, Chronic Wasting Disease oder Kuru). Andere Infektionswege wie etwa die Schmierinfektion konnten noch nicht ausgeschlossen werden. Pathogene Prionen können aber auch durch die spontane Umfaltung körpereigener Prionen entstehen (z. B. familiäre Variante der Creutzfeldt-Jakob-Krankheit, familiäre Schlaflosigkeit).
Auch in der heutigen modernen Medizin ist eine kurative Behandlung von Prionenerkrankungen nicht möglich, so dass Maßnahmen lediglich im palliativmedizinischen Rahmen möglich sind. Alle Krankheiten haben ebenfalls gemein, dass sie zu einem spongiformen (schwammartigen) Zerfall des Gehirns bzw. des vegetativen Nervensystems führen und somit grundsätzlich letal (tödlich) verlaufen. Die pathogene Prävalenz von durch Prionen verursachten Erkrankungen gilt gemein als extrem niedrig.
Grundsätzlich sind pathogene Prionen von anderen Krankheitserregern wie Viren, Bakterien oder Pilzen zu unterscheiden, da sie keine DNA oder RNA enthalten. Sie sind nicht nur von großem wissenschaftlichen und medizinischen Interesse, sondern hatten durch die „BSE-Krise“ auch starke Auswirkungen auf Gebiete wie Landwirtschaft, Verbraucherschutz und Politik.
Die vereinfachte Prionhypothese und Besonderheiten der Prionkrankheiten
Eines der zahlreichen im tierischen Körper vorkommenden Eiweiße heißt PrPC (Prion Protein cellular = zelluläres Prion-Protein). Es findet sich vor allem im Nervensystem, speziell im Gehirn. Zwischen den verschiedenen Tierarten und gegebenenfalls auch innerhalb einer Tierart unterscheiden sich die Prionen mehr oder weniger geringfügig. PrPC kommt vor allem an der Zelloberfläche vor und schützt die Zellen vor zweiwertigen Kupfer-Ionen, H2O2 und freien Radikalen. Des Weiteren wird vermutet, dass es einer der ersten Sensoren in der zellulären Abwehr von reaktivem Sauerstoff und freien Radikalen ist und Auswirkungen auf den enzymatischen Abbau von freien Radikalen hat.[3]
Gerät dieses normale Eiweiß PrPC in Kontakt mit einem PrPSc genannten Eiweiß (Prion Protein Scrapie; pathogene Form des Prion-Proteins, das in der Form zuerst bei an Scrapie erkrankten Tieren gefunden wurde), nimmt PrPC die Form von PrPSc an, „es klappt um“, es ändert seine Konformation. Es entwickelt sich eine Kettenreaktion, in der immer mehr PrPC in PrPSc umgewandelt werden. Große Mengen an PrPSc wirken zerstörerisch auf das Gehirn, da sie unlöslich sind und sich in den Zellen ablagern. Infolgedessen sterben diese Zellen ab; es entstehen Löcher im Gehirn, eine schwammartige Struktur entsteht. Daher auch der Name dieser Krankheit: spongiforme Enzephalopathie, schwammartige Gehirnerkrankung. Prionerkrankungen enden stets tödlich.
Die Initiation der Krankheit kann auf drei Weisen erfolgen, was unter allen Krankheiten einmalig ist:
- sporadisch, d. h. zufällig bzw. ohne erkennbare Ursache: PrPC faltet sich „zufällig“ in PrPSc um und löst dadurch die Kettenreaktion aus. Ein Beispiel ist die klassische Form der Creutzfeldt-Jakob-Krankheit (sCJD).
- genetisch, d. h. durch einen „Fehler“ im Erbmaterial: Das Gen PRNP, auf dem in Form von DNA die Information für die Herstellung von PrPC hinterlegt ist, kann eine Mutation enthalten. Das dann veränderte Protein ist anfälliger für eine Umwandlung in PrPSc. Die Mutation kann von den Eltern auf die Kinder vererbt werden. Beispiele sind die familiäre Form der Creutzfeldt-Jakob-Krankheit (fCJD), das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die tödliche familiäre Schlaflosigkeit (FFI Fatal Familial Insomnia: Letale familiäre Insomnie).
- durch Übertragung bzw. „Ansteckung“: Führt man sich von außen PrPSc zu, kann dies das eigene PrPC wiederum in PrPSc umwandeln, allerdings nicht unter allen Umständen. Es kommt darauf an, welche Menge zugeführt wird, in welcher Weise und um welche Art von PrPSc es sich genau handelt. Eine Ansteckung im alltäglichen Kontakt mit Patienten ist nicht möglich. Gut möglich ist dagegen eine Übertragung, wenn stark PrPSc-haltiges Material, also z. B. Gehirn von kranken Tieren oder Menschen, ins Blut oder schlimmstenfalls direkt ins Gehirn gelangt. Dies ist zum Beispiel bei der iatrogenen Form der Creutzfeldt-Jakob-Krankheit (iCJD) der Fall. Bei Operationen am Gehirn wurden durch nicht ausreichend sterilisierte Instrumente versehentlich Prionen von Erkrankten in das Gehirn von Gesunden gebracht. Ein anderes Beispiel ist Kuru, eine Krankheit auf Papua-Neuguinea; Angehörige des betroffenen Volksstamms führten sich dabei im Rahmen von kulturellen Riten das Gehirn von Verstorbenen und damit hohe Mengen an PrPSc zu. Das prominenteste Beispiel ist aber sicherlich die BSE („Rinderwahnsinn“). Nach der in der Wissenschaft als weitgehend gesichert anerkannten Theorie wurden diese PrPSc durch Tiermehl verbreitet, welches u. a. aus Kadavern von an Scrapie erkrankten Schafen hergestellt und dann an Rinder verfüttert wurde. Hinzu kam, dass man im Vereinigten Königreich die Verfahren zur Herstellung von Tiermehl so verändert hatte (niedrigere Temperatur bzw. Druck), dass diese PrPSc den Herstellungsprozess überstehen konnten. In geringerem Umfang haben sich wahrscheinlich auch andere Tiere (Katzen, Zootiere) durch dieses Tiermehl bzw. durch Verfütterung von Teilen von erkrankten Rindern infiziert. Schließlich erkrankten besonders in Großbritannien auch Menschen wohl aufgrund des Verzehrs von Fleisch bzw. Hirn- oder Rückenmarksgewebe von BSE-Kühen an einer neuen Form der Creutzfeldt-Jakob-Krankheit, der „neuen Variante“ (nvCJD oder vCJD). Schweizer Forscher entdeckten bei Versuchen mit Mäusen einen neuen Infektionsweg, über die Atemluft.[4] Ob eine solche Infektion auch beim Menschen möglich ist, ist noch umstritten.
Auch die familiären Formen von Prionkrankheiten lassen sich im Experiment übertragen, so kann beispielsweise das in einem Menschen aufgrund der genetischen Disposition entstandene PrPSc bei Mäusen die Krankheit auslösen, wenn es zuvor ins Gehirn gespritzt wurde.
Prionen sind sehr widerstandsfähig gegen übliche Desinfektions- bzw. Sterilisationsverfahren, was auch ein Grund für die iCJD-Fälle und die BSE-Krise war. Heute gibt es auf die erschwerte Inaktivierung von Prionen abgestimmte strenge Vorschriften für die Sterilisierung von Material, das mit möglicherweise prionhaltigem Gewebe in Kontakt gekommen ist. Da Infektionen oft erst bei der Obduktion entdeckt werden, muss ein Umgang mit Leichen immer so erfolgen, als läge eine Infektion vor.[5] Für den Umgang mit Leichen mit bestätigten Prionenerkrankungen sowie für Kontakt mit potentiell infektiösem Gewebe gelten aufgrund der hohen Resistenz der Erreger strengste Maßnahmen, die bis zur Exzision der betroffenen Stelle reichen.[6]
Die Prionhypothese gilt heute als relativ gesichert. Dass außer dem PrPSc noch ein weiterer Faktor eine Rolle spielt, kann jedoch noch nicht endgültig ausgeschlossen werden. Nachdem auch die intensive Suche nach Viren, Viroiden oder Nukleinsäure überhaupt erfolglos blieb, gibt es kaum noch Wissenschaftler, die diesen Weg weiter verfolgen. In der Öffentlichkeit kursieren gelegentlich Außenseitermeinungen wie etwa die Organophosphattheorie, nach der BSE im Zusammenhang mit Insektengiften steht, wofür es jedoch keine wissenschaftlichen Hinweise gibt.
Geschichte der Prionforschung
Einzelne Prionkrankheiten wurden schon vor langer Zeit beschrieben (Scrapie, die Prionkrankheit des Schafes, 1759 von Johann George Leopoldt; CJD 1920 von Hans Gerhard Creutzfeldt), ohne dass man etwas über die Ursache dieser Krankheiten wusste oder sie in eine Gruppe einordnen konnte. Nachdem 1932 die Übertragbarkeit von Scrapie nachgewiesen und 1957 erstmals Kuru beschrieben worden war, wurde Ende der 1950er Jahre von William J. Hadlow die Ähnlichkeit dieser Krankheiten festgestellt und Kuru ebenfalls experimentell auf Affen übertragen. Tikvah Alper und Mitarbeiter stellten 1966 fest, dass der Erreger zu klein war, um ein Virus zu sein[7], und offenbar keine Nukleinsäure enthielt[8][9]. Daher kam es zu einer "nur Protein"-Hypothese für den Erreger, wobei allerdings unklar blieb, wie sich so ein Protein vermehren könnte. Viele gingen daher am ehesten von Lentiviren als Ursache aus.
Die 1982 von Stanley Prusiner, der an Arbeiten von Daniel C. Gajdusek, welcher bereits unbewusst ein pathogenes Prion[10] entdeckt hatte, anknüpfte, veröffentlichte „Prionhypothese“ wurde zunächst in der Wissenschaft kritisch aufgenommen, da ein nukleinsäurefreies infektiöses Agens bis dahin nicht vorstellbar war. Im Nachhinein erwies sich diese Hypothese jedoch als bahnbrechend und 1997 wurde Prusiner für seine Arbeiten auf dem Gebiet der Prionforschung mit dem Nobelpreis geehrt. In den Jahren nach der Aufstellung dieser Hypothese konnten in zahlreichen Experimenten Hinweise für die Richtigkeit dieser Hypothese gewonnen werden, allerdings kein endgültiger Beweis. 1986 begann die BSE-Epidemie in Großbritannien, 1996 wurden die ersten Fälle von vCJD beschrieben.
Der Nachweis, dass rekombinantes Prion-Protein Krankheiten auslösen kann (womit das Koch'sche Postulat erfüllt war), gelang 2010[11].
Politiker versuchten, ihre Versäumnisse in Prävention und Verbraucherschutz unter anderem durch großzügige Ausgaben im Bereich der Prionforschung wettzumachen. Zahlreiche Arbeitsgruppen wurden neu eingerichtet, Zentren erbaut und Verbünde gegründet. Die Prionforschung wurde intensiviert und beschleunigt.[12]
2007 ergaben sich Zweifel, ob der Gehalt eines Gewebes an pathogenen Prionen in jedem Fall mit dessen Infektiosität korreliert.[13][14]
Neue Forschungsergebnisse um die US-amerikanische Forscherin Susan Lindquist zeigen, dass Prionen eine wichtige Rolle bei der Neurogenese (Entwicklung neuer Nervenzellen im Gehirn) spielen.
Struktur des Prion-Proteins
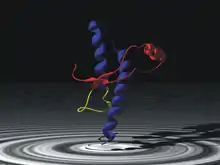
Physiologische (d. h. normale oder apathogene) Prionen haben zu 43 % die Struktur von Alpha-Helices. Die pathogenen Formen bestehen nur zu 30 % aus Alpha-Helices, zu 43 % bestehen sie aus Beta-Faltblatt-Strukturen. Die Gefahr der pathogenen Prionen besteht darin, dass sie in der Lage sind, die physiologischen, nicht pathogenen Prionen in pathogene umzuwandeln.
Das Prion-Protein ist ein beim Menschen aus 253 Aminosäuren (AS) bestehendes Glykoprotein, das im Prion-Protein-Gen (PRNP) codiert wird. Die AS-Homologie zu anderen Säugetieren beträgt 85 % oder mehr, zwischen Rind und Mensch gibt es z. B. 13 AS-Unterschiede. Es sind jeweils eine oder mehrere Mutationen bekannt, die zu fCJD, GSS oder FFI führen. Am Codon 129 besteht ein Methionin/Valin-Polymorphismus, der für Krankheitsausbruch und -verlauf mitentscheidend ist. PrPC enthält zum großen Anteil alpha-Helices, PrPSc mehr beta-Faltblattstrukturen, aber beide enthalten die gleiche Aminosäure-Primärsequenz.
Der genaue Vorgang der „Umfaltung“ von PrPC in PrPSc ist noch unbekannt. Diese verändert die Eigenschaften des Prion-Proteins; das PrPSc ist schlechter wasserlöslich, weil die hydrophoben Ketten nicht, wie bei der α-Helix üblich, zur Innenseite der Protein-Tertiärstruktur zeigen. Außerdem ist PrPSc weitestgehend resistent gegenüber vielen Desinfektionsmitteln, ionisierender und UV-Strahlung und hitzestabil. Feuchte Hitze (131 °C) in zur Sterilisation in der Medizin eingesetzten Autoklaven zerstört das PrPSc erst nach zwei Stunden, so dass medizinische Instrumente viermal hintereinander autoklaviert werden müssen, bei trockener Hitze wird das Prion bei 200 °C erst nach 60 Minuten inaktiviert und durch Proteasen nur schwer verdaulich (Proteasen können ein Protein am besten im entfalteten Zustand „zerschneiden“, die Denaturierung im Körper ist jedoch durch die veränderte Sekundärstruktur schlechter möglich). PrPC ist vor allem an Synapsen lokalisiert.
Die PrPC spielen laut neuester Erkenntnisse eine Rolle bei der Bildung von blutbildenden Stammzellen (s. u.). Prionprotein-Knockoutmäuse zeigen nach einem Schlaganfall eine verlangsamte Genesung, zudem neigen die Mäuse zu Fettleibigkeit. Es gibt jedoch Hinweise auf eine Rolle als kupferbindendes Protein an der Synapse. Laut einer Veröffentlichung von US-Forschern in der Fachzeitschrift PNAS erhalten normale Prion-Proteine die Regenerationsfähigkeit von blutbildenden Stammzellen. In diesen Zellen treten Prionen in der Zellmembran auf und erfüllen offenbar keine wichtigen Aufgaben – zumindest solange der Körper gesund ist.
Pathologie und Symptomatik von Prionkrankheiten
Prionkrankheiten sind vor allem durch motorische Störungen wie Ataxie und – am auffallendsten beim Menschen – kognitive Probleme bis zur Demenz gekennzeichnet. Nach einer Inkubationszeit von Jahren bis Jahrzehnten enden die Krankheiten stets tödlich. Im Gehirn finden sich bei der neuropathologischen Begutachtung unter dem Lichtmikroskop spongiöse (schwammartige) Veränderungen und, je nach Krankheit, unterschiedlich ausgeprägte Ablagerungen wie Amyloide, Kuru-Plaques und floride Plaques.
Auswahl aktueller Forschungsgebiete
- Funktion des PrPC
- genaue Struktur von PrPC und PrPSc
- Übertragungswege
- Blut-(Schnell-)Tests
- Therapieoptionen
- Möglichkeiten zur Prävention, Risikoabschätzung und Überwachung
Es wird angenommen, dass die Ansteckung mit der Prionenkrankheit durch Prion-assoziierte Proteine erfolgt und nicht durch das eigentliche Prionprotein. Von besonderem Interesse ist deshalb die Erforschung der Prion-assoziierten Proteine.
Meldepflicht
In der Schweiz ist der positive laboranalytische Befund zu «Prionen» für Laboratorien meldepflichtig und zwar nach dem Epidemiengesetz (EpG) in Verbindung mit der Epidemienverordnung und Anhang 3 der Verordnung des EDI über die Meldung von Beobachtungen übertragbarer Krankheiten des Menschen. Dies wird verstanden als «Positiver Nachweis von PrPSc in klinischem Material (insbesondere Gehirn)».[16]
Literatur
- Beat Hörnlimann, D. Riesner, H. Kretzschmar: Prionen und Prionenkrankheiten. de Gruyter, Berlin/ New York 2001, ISBN 3-11-016361-6.
- Martin H. Groschup: Prion diseases – diagnosis and pathogenesis. Springer, Wien 2000, ISBN 3-211-83530-X.
- Claudio Soto: Prions – the new biology of proteins. CRC, Taylor & Francis, Boca Raton 2006, ISBN 0-8493-1442-9.
- Sylvain Lehmann: Techniques in prion research. Birkhäuser, Basel 2004, ISBN 3-7643-2415-5.
- Andrew F. Hill: Prion protein protocols. Humana Press, Totowa 2008, ISBN 978-1-58829-897-3.
Weblinks
- Website Nationales Referenzzentrum für Prionerkrankungen, Georg-August-Universität Göttingen
- Ingrid Schütt-Abraham, Roland Heynkes: Theoretische TSE-Forschung. Website von Roland Heynkes
- prionforschung.de – Prion-Forschung · TSE-Koordinierungsstelle Niedersachsen
- Stefanie Offermann: Bluttest für Prionen: Forscher entwickeln neuen Diagnoseverfahren für die Eiweißerreger. In: wissenschaft.de. 29. August 2005, archiviert vom Original am 23. Februar 2006 (Hinweis auf Claudio Soto, Joaquín Castilla, Paula Saá: Detection of prions in blood. In: Nature Medicine 11/2005, 28. August 2005, S. 982–985, doi:10.1038/nm1286).
- Katharina Schöbi: Prionen auf verschlungenen Pfaden: Forscher: BSE-Erreger könnten ursprünglich vom Menschen stammen. In: wissenschaft.de. 2. September 2005, archiviert vom Original am 8. November 2005 (Hinweis auf den Artikel von Alan Colchester und Nancy Colchester in: The Lancet, Bd. 366, S. 856, 2005).
- Ilka Lehnen-Beyel: Wie Prionen von Schaf zu Schaf reisen: Studie zeigt, dass die infektiösen Eiweiße über den Urin übertragen werden können. In: wissenschaft.de. 14. Oktober 2005, archiviert vom Original am 25. Dezember 2005 (Hinweis auf den Artikel von Harald Seeger u. a. in: Science, Bd. 310, S. 324, 2005).
- Ilka Lehnen-Beyel: Der lange Schlaf der Prionen: Inkubationszeit kann mehr als fünfzig Jahre betragen. In: wissenschaft.de. 23. Juni 2006, archiviert vom Original am 3. Juli 2006 (Hinweis auf den Artikel von John Collinge u. a. in: The Lancet, Bd. 367, S. 2068).
- David S. Goodsell: Molecule of the Month: Prions. In: Protein Data Bank. Mai 2008, archiviert vom Original am 11. Mai 2008; abgerufen am 23. April 2018 (englisch).
Einzelnachweise
- Stanley B. Prusiner: Novel proteinaceous infectious particles cause scrapie. Science 1982, 216(4542), S. 136–144. PMID 6801762.
- Das Wort prion wird auch als Abkürzung für den von Prusiner verwendeten Begriff proteinaceous infectious particle dargestellt, vgl. proteinaceous im LEO-Wörterbuch
- Vgl. Reactive Oxygen Species-mediated β-Cleavage of the Prion Protein in the Cellular Response to Oxidative Stress. In: Journal of Biological Chemistry. Vol. 280, NO.43, S. 35914–35921.
- Vgl. Haybaeck et al. Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. In: PLoS Pathog. 7(1), S. e1001257, 2011. doi:10.1371/journal.ppat.1001257
- Hygieneplan Pathologie - Obduktionsbereich. Medizinische Universität Wien, abgerufen am 20. April 2021.
- Hygieneplan Sondersektion Klinische Pathologie. Medizinische Universität Wien, abgerufen am 20. April 2021.
- Tikvah Alper, D.A. Haig, M.C. Clarke: The exceptionally small size of the scrapie agent. In: Biochemical and Biophysical Research Communications. Band 22, Nr. 3, S. 278–284, doi:10.1016/0006-291x(66)90478-5 (elsevier.com [abgerufen am 31. Oktober 2017]).
- Tikvah Alper, W. A. Cramp, D. A. Haig, M. C. Clarke: Does the Agent of Scrapie Replicate without Nucleic Acid ? In: Nature. Band 214, Nr. 5090, 20. Mai 1967, S. 764–766, doi:10.1038/214764a0 (nature.com [abgerufen am 31. Oktober 2017]).
- R. Latarjet, B. Muel, D. A. Haig, M. C. Clarke, Tikvah Alper: Inactivation of the Scrapie Agent by Near Monochromatic Ultraviolet Light. In: Nature. Band 227, Nr. 5265, 26. September 1970, S. 1341–1343, doi:10.1038/2271341a0 (nature.com [abgerufen am 31. Oktober 2017]).
- Gisela Baumgart: Prusiner, Stanely Ben. In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin/ New York 2005, ISBN 3-11-015714-4, S. 1188.
- Fei Wang, Xinhe Wang, Chong-Gang Yuan, Jiyan Ma: Generating a Prion with Bacterially Expressed Recombinant Prion Protein. In: Science. Band 327, Nr. 5969, 26. Februar 2010, ISSN 0036-8075, S. 1132–1135, doi:10.1126/science.1183748, PMID 20110469 (sciencemag.org [abgerufen am 31. Oktober 2017]).
- Stephan Schröder-Köhne: Fortschritte in der Prionenforschung. Vom 21. April 2005.
- R. M. Barron u. a.: High titres of TSE infectivity associated with extremely low levels of PrPSc in vivo. In: J Biol Chem. 2007 Oct 8. PMID 17923484
- P. Piccardo u. a.: Accumulation of prion protein in the brain that is not associated with transmissible disease. In: Proc Natl Acad Sci U S A. 2007 Mar 13;104(11), S. 4712–4717. PMID 17360589.
- Protein Data Bank: Human Prion Protein Fragment 121-230
- Meldepflichtige übertragbare Krankheiten und Erreger. (PDF, 4 MB) Leitfaden zur Meldepflicht 2020. Bundesamt für Gesundheit BAG, Abteilung Übertragbare Krankheiten, 23. Februar 2020, S. 18, abgerufen am 8. März 2020.