Granularzelltumor
Der Granularzelltumor (Granularzellmyoblastom, Myoblastenmyom, Abrikossow-Tumor) ist ein seltener gutartiger nichtepithelialer Tumor vermutlich neuroektodermaler Abstammung, der sich meist im mittleren Lebensalter manifestiert. Hauptlokalisationen sind die Zunge sowie Haut und Unterhaut des Körperstammes. Daneben kann der Tumor an praktisch jeder anatomischen Lokalisation auftreten. Klinisch handelt es sich um einen langsam wachsenden, in der Regel schmerzlosen Tumor, der nach chirurgischer Entfernung nur selten rezidiviert. Eine maligne Entartung wird in einem kleinen Prozentsatz der Fälle beobachtet.[1]
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9580/0 | Granularzelltumor |
| 9580/3 | Maligner Granularzelltumor |
| ICD-O-3 erste Revision online | |
Geschichtliches
Die Erstbeschreibung des Granularzelltumors erfolgte bereits 1926 durch Alexei Iwanowitsch Abrikossow, der die Läsion zunächst als eine gutartige Neoplasie der quergestreiften Skelettmuskulatur interpretierte und mit der Bezeichnung Myoblastenmyom belegte.[2][3]
Ätiologie
Die dem Granularzelltumor zugrunde liegenden Ursachen sind unbekannt. Als wahrscheinlicher Ausgangspunkt der Tumorentstehung gilt die Schwann-Zelle, wobei allerdings eine Beziehung nicht in allen Fällen hergestellt werden kann.[4] Die seltene angeborene Variante des gingivalen Granularzelltumors ist möglicherweise eine nicht-neoplastische, reaktive Läsion.[1]
Epidemiologie
Der Altersgipfel der Erkrankung liegt im mittleren Lebensalter, wobei der Manifestationszeitpunkt jedoch in weiten Grenzen variiert. Frauen sind etwas häufiger betroffen als Männer.[1]
Pathologie
_skin.jpg.webp)
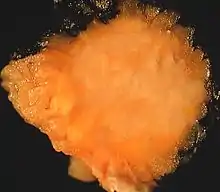
Makroskopisch handelt es sich um meist kleine, üblicherweise weniger als 3 cm messende, blass gelbliche, häufig unscharf begrenzte Tumoren von fester Konsistenz, die bevorzugt in der Zunge (40 %) sowie in Haut und Unterhaut insbesondere des Körperstammes (30 %) auftreten. Darüber hinaus wurden Granularzelltumoren an vielen weiteren Lokalisationen beschrieben, darunter Bronchialsystem (13 %), Harn- und Geschlechtsapparat (13 %), Magen-Darm-Trakt (6 %) oder Zentralnervensystem.[1][4][5]
Intrakraniell tritt der Tumor vor allem in der Hypophysenregion auf und wird hier als Granularzelltumor der Neurohypophyse bezeichnet, der nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems als Grad I klassifiziert wird.[6]
Histologisch zeigen Granularzelltumoren unabhängig von ihrer Lokalisation ein bemerkenswert uniformes Erscheinungsbild. Die Tumorzellen sind in Nestern oder Zellballen angeordnet, groß, rund, polygonal oder elongiert und besitzen reichlich feingranuläres, eosinophiles Zytoplasma, in dem sich zuweilen größere eosinophile Tröpfchen oder Granula finden. Diese enthalten reichlich hydrolytische Enzyme wie die saure Phosphatase und lassen sich regelmäßig mit dem Farbstoff Luxol Fast Blue, in einem Teil der Fälle auch in der PAS-Färbung anfärben. Die Zellgrenzen sind häufig indistinkt, was zum Eindruck eines synzytialen Zellverbandes führen kann.[4] Die Zellkerne sind klein, zentral lokalisiert und meist pyknotisch oder hyperchromatisch, seltener auch vesikulär. Mitosen und geringfügige, oft degenerativ bedingte Atypien werden nur selten beobachtet. Häufig werden Tumorzellgruppen in Umgebung kleiner Nerven gefunden. Oberflächlich lokalisierte Tumoren sind oft begleitet von einer pseudoepitheliomatösen Hyperplasie des überkleidenden Plattenepithels, die nicht mit einem Plattenepithelkarzinom verwechselt werden darf.[1]
Immunhistochemie
_S-100.JPG.webp)
Immunhistochemisch zeigen die Tumorzellen des Granularzelltumors eine Positivität für Neuronenspezifische Enolase (NSE), CD63 (NK1-C3), S-100 sowie fast immer auch für Inhibin und Calretinin. Außerdem besteht eine feingranuläre Positivität für das lysosomale Antigen CD68.[1]
Die seltenen malignen Granularzelltumoren zeigen häufig eine negative Immunoreaktivität für NSE, S-100 und Vimentin.[7]
Diagnose und Differentialdiagnose

Die Diagnose erfolgt nach Entnahme einer Gewebeprobe (Biopsie) oder am Präparat des vollständig entfernten Tumors durch den Pathologen. In der Regel ist das histologische Bild so charakteristisch, dass keine diagnostischen Probleme auftreten. Differentialdiagnostisch in Frage kommen je nach Lokalisation das Schwannom, Neurofibrom, das alveoläre Weichteilsarkom, das adulte Rhabdomyom, das histiozytoide Karzinom, das Leiomyom oder der gastrointestinale Stromatumor sowie selten auch reaktive Läsionen nach vorausgegangenem Trauma oder Entzündung.[4][1]
Therapie
Therapie der Wahl ist die chirurgische Entfernung des Tumors. Ein weiter Sicherheitsabstand zum Tumor ist dabei nur bei der malignen Variante des Granularzelltumors erforderlich.[7]
Prognose
Als üblicherweise benigne Neoplasie mit langsamem Wachstum zeigt der Granularzelltumor eine gute Prognose. Die Rezidivquote nach chirurgischer Therapie liegt bei unter 5 Prozent; ein Wiederauftreten des Tumors ist dabei in der Regel auf eine unvollständige Entfernung zurückzuführen. Eine maligne Entartung wird in höchstens 2–3 Prozent der Fälle beobachtet. Hierbei kommt es im Verlauf häufig zu einer Metastasierung mit letztendlich tödlichem Ausgang.[1]
Einzelnachweise
- C. D. M. Fletcher: Diagnostic Histopathology of Tumors. 3. Auflage. Churchill Livingstone, 2007.
- A. Abrikossoff: Über Myome, ausgehend von der quergestreiften willkürlichen Muskulatur. In: Arch Pathol Anat. 1926;260, S. 214.
- A. Abrikossoff: Weitere Untersuchungen über Myoblastenmyome. In: Arch Pathol Anat. 1931;280, S. 723.
- PathConsult: Granular Cell Tumor. (24. Februar 2006), Elsevier; http://www.pathconsultddx.com/pathCon/diagnosis?TXTBOX2=gra&pii=S1559-8675%2806%2970247-9@1@2Vorlage:Toter+Link/www.pathconsultddx.com (Seite+nicht+mehr+abrufbar,+Suche+in+Webarchiven) Datei:Pictogram+voting+info.svg Info:+Der+Link+wurde+automatisch+als+defekt+markiert.+Bitte+prüfe+den+Link+gemäß+Anleitung+und+entferne+dann+diesen+Hinweis.+
- T. Schlick, T. Junginger: Abrikossoff-Granulosazelltumor: Ein seltener Tumor des Oesophagus. In: Chirurg. 1997 Sep;68(9), S. 932–935. PMID 9410685
- Cohen-Gadol u. a.: Granular Cell Tumor of the Sellar and Suprasellar Region: Clinicopathologic Study of 11 Cases and Literature Review. In: Mayo Clin Proc. 2003;78(5), S. 567–573. PMID 12744543 Volltext (Seite nicht mehr abrufbar, Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.
- The Maxillofacial Center for Education & Research: Granular Cell Tumor; Archivlink (Memento des Originals vom 26. Januar 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
 Datei:Pictogram+voting+info.svg Info:+Der+Link+wurde+automatisch+als+defekt+markiert.+Bitte+prüfe+den+Link+gemäß+Anleitung+und+entferne+dann+diesen+Hinweis.+){kind=link}