Paroxysmale nächtliche Hämoglobinurie
Die paroxysmale nächtliche Hämoglobinurie (PNH) (synonym: Marchiafava-Micheli-Syndrom) ist eine seltene, potenziell lebensbedrohliche Erkrankung des Blutes, bei der es durch einen erworbenen Gendefekt zur Zerstörung vor allem roter Blutkörperchen (Erythrozyten) durch einen Teil des Immunsystems, das Komplementsystem, kommt.
| Klassifikation nach ICD-10 | |
|---|---|
| D59.5 | Paroxysmale nächtliche Hämoglobinurie [Marchiafava-Micheli], exkl. Hämoglobinurie o.n.A. |
| ICD-10 online (WHO-Version 2019) | |
Die PNH kann durch eine Anämie mit Kurzatmigkeit und schnellem Herzschlag, Thromboseneigung, starke Erschöpfung, Bauch- und Rückenschmerzen und eine Dunkelfärbung des Urins durch Hämoglobin (Hämoglobinurie) auffallen. Die Symptome können sich anfallsartig (paroxysmal) verschlimmern. Die namensgebende nächtlich auftretende Hämoglobinurie tritt nur bei einem Viertel der Patienten auf. Der Verlauf kann sehr unterschiedlich sein, die Krankheit ist unbehandelt aber häufig tödlich. Die häufigste Todesursache sind Thrombosen, häufig an untypischen Stellen wie den Lebervenen oder am Gehirn (Sinusthrombose).
Die Ursache liegt im Knochenmark, wo aus blutbildenden Stammzellen die Blutzellen gebildet werden. Bei Menschen mit PNH liegt in einer oder mehreren blutbildenden Stammzellen ein Defekt des PIGA-Gens vor, wodurch ein bestimmter Stoff nicht mehr gebildet werden kann: das Glycolipid GPI. GPI ist ein sogenannter Proteinanker, der verschiedene Proteine an der Zelloberfläche befestigt. Unter diesen Proteinen sind zwei wichtige Proteine (CD55 und CD59), die sonst die Blutzellen vor dem Angriff des Komplementsystems schützen. Daher werden alle Blutzellen ohne GPI vom Komplementsystem attackiert, die roten Blutkörperchen sind aber sehr viel anfälliger als die anderen Zellen.
Die PNH ist häufig ein Begleitphänomen der aplastischen Anämie, bei der es zum allgemeinen Rückgang der Stammzellen im Knochenmark kommt. Vermutlich liegt dem eine Autoimmunreaktion gegen die normalen Stammzellen mit GPI zugrunde, wodurch sich die Stammzellen mit GPI-Defekt ausbreiten können. Die Vorgänge im Knochenmark sind Gegenstand der aktuellen Forschung.
Der Greifswalder Arzt Paul Strübing beschrieb 1882 den ersten Fall eines Patienten mit paroxysmaler nächtlicher Hämoglobinurie. Die Erforschung der paroxysmalen nächtlichen Hämoglobinurie führte zur Entdeckung des alternativen Wegs der Komplementaktivierung und dadurch zu einem besseren Verständnis der humoralen Immunantwort. Gleichzeitig ist die Erkrankung ein Beispiel dafür, wie Forschung sowohl von sorgfältigen Experimenten als auch von glücklichen Zufällen abhängt.
Bis 2007 konnte die Erkrankung nur unterstützend behandelt werden, z. B. mit Bluttransfusionen. Für schwere Fälle stand als letztes Mittel die Knochenmarktransplantation zur Verfügung. Seit 2007 ist mit Eculizumab eine zielgerichtete Therapie der Erkrankung möglich. Der Wirkstoff hemmt den Angriff des Komplementsystems.
Krankheitsbild
Epidemiologie
Die Erkrankung ist nicht vererbbar. Eine Häufung unter Verwandten oder eine spezielle Geschlechterverteilung wurde bisher nicht beobachtet,[1] allerdings sind in einer großen Beobachtungsstudie mit vornehmlich weißen Patienten aus westlichen Industriestaaten die Frauen mit 54 % leicht in der Mehrheit.[2] Die Prävalenz wird auf ca. 16/1 Million Einwohner geschätzt, und die Rate der Neuerkrankungen wird nach Zahlen für Großbritannien und Frankreich auf ca. 1,3/1 Million pro Jahr beziffert.[3] Gehäuft bricht die Krankheit im Alter von 25 bis 45 Jahren aus.[1] Weltweit liegt die Inzidenz zwischen 1 und 1,5 Neuerkrankungen pro 1 Million Einwohner pro Jahr. In Asien tritt die PNH dabei häufiger auf als in anderen Teilen der Welt.[4]
Symptome
.svg.png.webp)
Die Ausprägung der Symptome ist von Patient zu Patient sehr unterschiedlich. Häufig fällt die Erkrankung zunächst durch die hämolytische Anämie auf, in vielen Fällen ist sie kombiniert mit einer aplastischen Anämie durch das Knochenmarkversagen. Viele Patienten haben bereits eine allgemeine Verringerung aller Blutzellen (Panzytopenie) als Ausdruck des Knochenmarkversagens. Die namensgebende anfallsartig auftretende (paroxysmale), nächtliche Ausscheidung von Hämoglobin in den Urin (Hämoglobinurie) mit dunkel gefärbtem Urin tritt nur bei etwa jedem vierten[5] Patienten auf. Das dritte Kardinalsymptom neben Anämie und Panzytopenie ist eine ausgeprägte Blutgerinnungsneigung (Thrombophilie). Viele Patienten leiden unter schwerer Erschöpfung (Fatigue). Sie hängt nicht mit der Anämie zusammen, sondern vom Ausmaß der Hämolyse-Aktivität. Sie wird häufig durch Infekte, körperliche Anstrengung, Operationen und Schwangerschaften verschlimmert. Manche Patienten klagen über Bauch- und Rückenschmerzen, Speiseröhrenkrämpfe, Schluckbeschwerden und Erektionsstörungen. Bei den meisten Patienten bestehen oder entwickeln sich Einschränkungen der Nierenfunktion sowie ein erhöhter Blutdruck in den Lungenarterien (Pulmonale Hypertonie).[4]
Zu diesen Symptomen, die auf die Hämolyse zurückgeführt werden können, kommen die Beschwerden durch die Anämie: Schwindel, Kopfschmerzen, Kurzatmigkeit, schneller Herzschlag und Blässe. Je nachdem, wie weit das Knochenmarkversagen fortgeschritten ist, kommen die Folgen durch die Panzytopenie hinzu: Infektanfälligkeit durch den Mangel an Immunzellen und eine Blutungsneigung durch den Mangel an Blutplättchen.[5]
Für Patientinnen mit PNH stellt eine Schwangerschaft eine erhebliche Gefahr dar. Das Thromboserisiko erhöht sich durch die Schwangerschaft nochmals, wodurch Schwangere eine erhöhte Sterblichkeit im Vergleich zu anderen PNH-Patienten aufweisen und mehr Fehlgeburten als gesunde Frauen erleiden.[3]
Verlauf und Prognose
Der natürliche Verlauf der PNH kann sich über Jahre hinziehen. Noch in den 1990er Jahren verstarb die Hälfte der Erkrankten innerhalb von 10 Jahren nach Diagnosestellung.[6] In den frühen 2000er Jahren verlängerte sich diese Zeit auf 20 Jahre.[7] Patienten unter Eculizumab-Langzeittherapie (s. u.) können die gleiche Lebenserwartung wie Gesunde haben.[8]
Vor Einführung wirksamer Therapien waren Thrombosen die Haupttodesursache bei PNH-Patienten. Die Thrombosen treten meistens an den Lebervenen (Budd-Chiari-Syndrom) auf, andere häufig betroffene Venen sind die Pfortader, die Mesenterialvenen, die Milzvene und die venösen Sinus des Gehirns (Sinusthrombose). Auch die tiefen Venen der Beine und Arme können betroffen sein, wodurch es zu Lungenembolien kommt. Arterielle Thrombosen sind seltener und können unter anderem Herzinfarkte und Schlaganfälle verursachen. Paroxysmale nächtliche Hämoglobinurie tritt regelhaft mit einem Knochenmarksversagen durch eine Autoimmunreaktion auf. Bei Patienten mit ausgeprägter aplastischer Anämie führt langfristig das Knochenmarksversagen zum Tod.[4] Bei etwa ein bis zwei Prozent der Erkrankten geht die PNH in eine akute myeloische Leukämie über.[5]
Krankheitsentstehung
Pathogenese
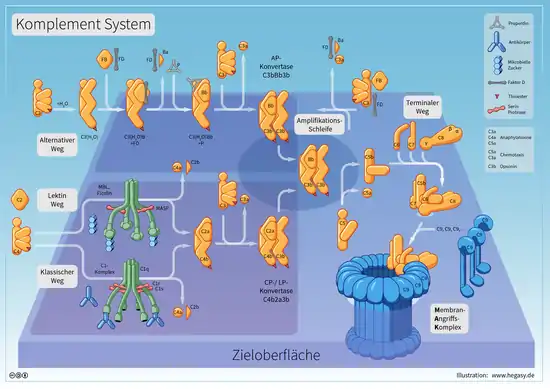
Das Komplementsystem besteht aus verschiedenen Proteinen im Blut, deren Aufgabe es ist, eingedrungene Mikroorganismen wie Bakterien zu erkennen, ihre Zellmembran zu eröffnen und dadurch zu zerstören (lysieren). Die Blutzellen des Körpers werden verschont, weil bestimmte Proteine auf der Oberfläche der Zellen, CD55 und CD59, die Attacke des Komplementsystems verhindern. Die Proteine müssen allerdings mit einem Glycolipid an der Zellmembran verankert werden, dieser Anker heißt Glycosylphosphatidylinositol, kurz GPI. Bei PNH fehlt vielen Blutzellen GPI, weswegen sie ohne CD55 und CD59 den Angriffen des Komplementsystems ausgesetzt sind. Die roten Blutkörperchen (Erythrozyten) sind am anfälligsten für die Zerstörung, weil sie keinen Zellkern haben.[4]
Die Ursache liegt in den blutbildenden Stammzellen des Knochenmarks. Bei der PNH tritt in einer oder mehreren Stammzellen ein erworbener Defekt (somatische Mutation) in dem Gen PIGA auf. PIGA ist das Gen für das Enzym N-Acetylglukosaminyltransferase. Dieses ist für den ersten Schritt der GPI-Synthese zuständig und verliert durch die Mutation seine Funktion. Theoretisch kann durch den Verlust jedes der über 20 an der GPI-Herstellung (Synthese) beteiligten Gene die PNH entstehen. Praktisch gibt es nur einen einzigen bekannten Fall, in dem nicht eine PIGA-Mutation die Ursache war. Das liegt daran, dass PIGA auf dem X-Chromosom liegt. Männer haben nur ein X-Chromosom, Frauen haben zwar zwei, davon ist jedoch nur eines aktiv, das andere ist „abgeschaltet“. Deswegen reicht eine einzige Mutation in PIGA, um die GPI-Synthese zu verhindern. Alle anderen Gene liegen doppelt vor, es müssten also beide Varianten (Allele) des gleichen Gens durch eine Mutation ausgeschaltet sein, um die GPI-Synthese zu beeinträchtigen. Diese Konstellation ist aber unwahrscheinlich und daher selten.[4]
Zum Ausbruch der Krankheit reicht die PIGA-Mutation allein nicht aus, da auch bei den meisten gesunden Menschen ohne PNH PIGA-mutierte Stammzellen nachgewiesen werden können. Es muss also Faktoren geben, durch welche die mutierte Stammzelle zwischen den anderen, nicht mutierten, Stammzellen im Knochenmark die Vorherrschaft erlangt. Hier wird einerseits eine Autoimmunreaktion gegen gesunde, GPI-tragende Stammzellen diskutiert. Dadurch ist das häufig zu beobachtende gleichzeitige Auftreten von PNH und Knochenmarkversagen (aplastische Anämie) zu erklären. Durch den Schwund gesunder Stammzellen erhalten die GPI-losen Stammzellen Platz zur Ausbreitung. Andererseits zeigten Genom-Studien, dass in den PIGA-mutierten Stammzellen bei PNH noch andere Mutationen in Genen auftreten, die für die Regulation des Zellwachstums eine Rolle spielen und häufig auch bei Blutkrebserkrankungen gefunden werden.[4]
Anämie und Hämolyse
Das zentrale Zeichen der PNH ist die ständige Hämolyse durch das Komplementsystem. Sie verstärkt sich durch bestimmte Auslöser wie Infektionen, weil dann über Antikörper das Komplementsystem verstärkt aktiviert wird, und im Schlaf. Das durch die Hämolyse freigesetzte Hämoglobin wird über die Nieren ausgeschieden und bewirkt eine Dunkelfärbung des Urins.[4] Der Zusammenhang zwischen Schlaf und Hämolyse beschäftigte lange die Forschung. Die gängige Vermutung ist, dass es im Schlaf zu einer leichten Hypoventilation kommt. Dadurch wird weniger CO2 abgeatmet, das sich folglich im Blut anreichert. Dadurch sinkt der pH-Wert, das Blut wird also saurer, was die Aktivität des Komplementsystems steigert. Diese Hypothese wurde nie bewiesen, zumal nur eine Minderheit der Patienten die klassische „nächtliche“ Hämoglobinurie zeigt. Nicht alle PNH-Erythrozyten sind gleich empfindlich gegenüber den Angriffen des Komplementsystems. Je nach zugrundeliegendem Defekt in PIGA können die betroffenen Zellen in unterschiedlichem Ausmaß GPI-verankerte Proteine auf ihrer Zellmembran zeigen. Die Stärke der Hämolyse hängt dann davon ab, wie viel (oder wie wenig) CD55 und CD59 auf der Zelloberfläche verbleibt.[9]
Knochenmarkversagen
Die PNH ist mit anderen Knochenmarkserkrankungen assoziiert. Etwa 20 % der PNH-Patienten haben zum Zeitpunkt der Diagnosestellung auch Zeichen eines allgemeinen Rückgangs der Blutbildung im Knochenmark durch eine aplastische Anämie.[4] Zum besseren Verständnis dieses Zusammenhangs hilft es, sich die Ursache der aplastischen Anämie vor Augen zu führen: Bei der plötzlich auftretenden Form der aplastischen Anämie bei jüngeren Menschen liegt in der Regel ein Angriff zytotoxischer T-Zellen auf das eigene Knochenmark vor. Was diese Autoimmunreaktion auslöst, ist nicht klar, wahrscheinlich gibt es mehrere Auslöser und Zielstrukturen. Bei etwas mehr als jedem zehnten Fall „entkommen“ aber Knochenmarkzellen der Attacke. Es sind mehrere Eigenschaften von Knochenmarkzellen bekannt, die dazu führen, dass sie von den angreifenden T-Zellen verschont werden. Sind also die PNH-Stammzellen ohne GPI vor der Autoimmunreaktion geschützt, liegt der Schluss nahe, dass diese etwas mit dem GPI-Anker zu tun haben muss – bis heute ist aber kein Antigen identifiziert worden.[10]
Dystonie glatter Muskelzellen und Thrombosen
Einige Symptome wie die Schmerzen, Schluckstörungen, die Nierenfunktionseinschränkungen und die pulmonale Hypertonie können durch Anspannung der glatten Muskelzellen, die in den Wänden von Blutgefäßen und der Verdauungsorganen vorkommen, erklärt werden. Sie sind eine Folge der Hämolyse. Normalerweise wird freies Hämoglobin im Blut über verschiedene Mechanismen eliminiert. Bei den hämolytischen Anfällen fällt jedoch so viel Hämoglobin an, dass die Schutzmechanismen überfordert sind.[4] Dieses freie Hämoglobin bindet dann Stickstoffmonoxid (NO) in den Gefäßen. Stickstoffmonoxid sorgt für eine Entspannung glatter Muskelzellen, fehlt es, kontrahieren die glatten Muskelzellen.[11] Blutgefäße verengen sich dadurch, was die Erektions- und Nierenstörungen sowie den Bluthochdruck in der Lunge erklärt. Auch in den Wänden des Verdauungstraktes kontrahieren Muskelzellen, daher die Bauchkrämpfe und Schluckstörungen.[4]
Der Entstehungsmechanismus der Thrombosen ist unklar und ein Problem für die zukünftige Forschung. Eine Möglichkeit ist die erhöhte Thrombozytenaktivität durch den Stickstoffmonoxid-Mangel, der auch mit der Dystonie glatter Muskelzellen in Verbindung gebracht wird.[4]
Einteilung
Aufgrund der Verknüpfung der PNH mit anderen Knochenmarkerkrankungen gibt es vielfältige Ausprägungen der Krankheit. Es gibt zwei Klassifikationen, die versuchen, dies abzubilden, durchgesetzt hat sich bislang keine.[4] Die ältere von 2005[12] unterscheidet drei Unterkategorien: 1. die PNH mit Hämolyse und Thrombosen. 2. die PNH im Kontext einer anderen Knochenmarkerkrankung (aplastische Anämie, Myelodysplastisches Syndrom). 3. die subklinische PNH. Hier haben Patienten geringe Anteile an PNH-Zellen, aber keine Symptome. Die neuere von 2008[7] ist mittlerweile verbreiteter.[2][4] Auch sie unterscheidet drei Unterkategorien: 1. die hämolytische (klassische) PNH. Es liegen Thrombosen oder eine Hämolyse vor. 2. die aplastische-Anämie-PNH (AA-PNH) mit zusätzlichen Hinweisen auf ein Knochenmarkversagen. Diese beiden Gruppen sind über Grenzwerte der Hämoglobin-Konzentration, der Anzahl bestimmter Immunzellen und der Blutplättchen definiert. Patienten, die aufgrund ihrer Blutwerte keiner der beiden Unterkategorien zugeordnet werden, fallen daher in Kategorie 3: Intermediate PNH, deutsch etwa „mittlere PNH“.
Diagnose
Diagnostik
Die Diagnosestellung wird wesentlich von den Symptomen des Patienten geleitet. Häufig ergibt sich der Verdacht auf eine Paroxysmale nächtliche Hämoglobinurie bei Patienten, bei denen eine Blutarmut aufgefallen ist.[5]
Bei der Laboruntersuchung des Blutes zeigen sich dann Zeichen einer hämolytischen Anämie: Der Hämoglobinwert und die Zahl der roten Blutkörperchen sind erniedrigt. Gleichzeitig ist die Konzentration der Laktatdehydrogenase (LDH) erhöht, weil sie aus den zerstörten roten Blutkörperchen freigesetzt wird. Unkonjugiertes Bilirubin (ein Abbauprodukt von Hämoglobin) ist ebenfalls erhöht. Das Haptoglobin, das freies Hämoglobin im Blut bindet, ist erniedrigt oder nicht nachweisbar. In der Regel wird auch ein Blutausstrich durchgeführt. Dieser erlaubt es, das Aussehen der Erythrozyten zu beurteilen. Insbesondere finden sich bei der PNH keine Fragmentozyten, wie sie bei anderen hämolytischen Anämien auftreten (s. Differentialdiagnose).[3]
Der Ham-Test wird heute nicht mehr angewendet. Bei diesem wird das Patientenblut leicht angesäuert, bei PNH kommt es zur Hämolyse.[13] Ein weiterer nicht mehr verwendeter Test ist der Sucrose-Lyse-Test. Hier wird durch Zugabe von Saccharose das Komplementsystem aktiviert.[14] Beide Tests waren nicht hinreichend aussagekräftig.[15] Stattdessen wird zum sicheren Nachweis der Erkrankung die Durchflusszytometrie einer peripheren Blutprobe angewendet: Mit dieser Untersuchung kann das Fehlen von GPI-verankerten Proteinen entdeckt werden. Gibt es Blutzellen aus mindestens zwei verschiedenen Blutzellreihen (z. B. Erythrozyten und Granulozyten), denen mindestens zwei solcher Proteine fehlen, gilt die PNH als sehr wahrscheinlich.[1][4] Eine neuere Methode weist GPI direkt nach: Fluorescence-labeled Aerolysin (FLAER).[4] Aerolysin ist ein Toxin des Bakteriums Aeromonas hydrophilius, das unmittelbar an den GPI-Anker bindet. Es ist mit einem Fluoreszenz-Farbstoff versehen, der unter dem Mikroskop erkannt werden kann (Fluoreszenzmikroskopie). Zellen, denen GPI fehlt, färben sich deswegen nicht an.[16]
Differentialdiagnosen
Die wichtigste Differentialdiagnose zur PNH ist die Autoimmunhämolytische Anämie. Bei dieser bildet das Immunsystem Antikörper gegen die roten Blutkörperchen. Diese Antikörper lassen sich im Coombs-Test nachweisen. Bei PNH bleibt der Coombs-Test negativ, da keine Antikörper gegen rote Blutkörperchen vorliegen (Coombs-negative hämolytische Anämie).[1][12] Andere Differentialdiagnosen sind angeborene hämolytische Anämien, die sich meistens schon im Kindesalter zeigen, sowie andere erworbene hämolytische Anämien:
- Angeborene hämolytische Anämien:
- Hämoglobinopathien: z. B. Sichelzellanämie, Thalassämie
- Erythrozyten-Enzym-Defekte: z. B. Glucose-6-phosphat-Dehydrogenase-Mangel
- Erythrozyten-Membran-Defekte: z. B. Kugelzellanämie, hereditäre Elliptozytose
- familiäres Hämolytisch-urämisches Syndrom
- angeborene Thrombotisch-thrombozytopenische Purpura (=Upshaw-Schulman-Syndrom)
- Erworbene hämolytische Anämien:
- Autoimmunhämolytische Anämie
- Mechanische Zerstörung: z. B. durch mechanische Herzklappen, Marschhämoglobinurie
- Verbrennung
- Gifte: Schlangengifte, Metalle, bestimmte Medikamente
- Zieve-Syndrom
Therapie
Die Therapieempfehlungen für den deutschsprachigen Raum werden in Zusammenarbeit der deutschen, österreichischen und Schweizer Gesellschaften für Hämatologie auf onkopedia.com veröffentlicht (s. Weblinks). Generell erfolgt die Behandlung der zumeist vorliegenden Anaplastischen Anämie nach der Leitlinie zur Anaplastischen Anämie. Die Therapie der Hämolyse-bezogenen Krankheitszeichen richtet sich nach der Ausprägung der Symptome. Bei hämolytischer Anämie kann Eisen und Vitamin B12 zur Unterstützung der Blutneubildung verabreicht werden. Bei schweren Blutverlusten muss gegebenenfalls Blut transfundiert werden. Haben Patienten Thrombosen, benötigen sie eine lebenslange Hemmung der Blutgerinnung, beispielsweise mit Marcumar, Heparin oder DOAKs. Diese Therapie schließt erneute Thrombosen im Fall der PNH allerdings nicht aus. Bakterielle Infektionen sollen früh mit Antibiotika behandelt werden, um hämolytische Anfälle zu vermeiden.[3]
Lange war die einzige Aussicht auf Heilung eine Knochenmarktransplantation. Diese Therapie beinhaltet für den Patienten allerdings ein großes Risiko, zu versterben, weswegen sie nur bei schweren Fällen als letztes Mittel in Frage kam.[4]
1996 wurde Eculizumab vorgestellt:[17] Der gentechnisch hergestellte monoklonale Antikörper gegen den Komplementfaktor C5 wurde in den Folgejahren in mehreren klinischen Studien getestet.[18][19][20] Seit 2007 ist Eculizumab in den USA und in der EU unter dem Markennamen Soliris zugelassen. Studienergebnisse mit dem Wirkstoff zeigten eine schnelle und anhaltende Kontrolle der chronischen Hämolyse. PNH-Patienten erleiden unter Eculizumab-Therapie fast keine Thrombosen mehr, was sonst die Haupttodesursache darstellte.[8][21] Fast die Hälfte der Patienten benötigte keine Bluttransfusionen mehr.[18] Auch die für eine PNH typischen Symptome wie z. B. Bauchschmerzen, Schluckbeschwerden (Dysphagie) und Potenzstörungen (erektile Dysfunktion) konnten mit einer Eculizumab-Behandlung verbessert werden.[19] Insgesamt zeigte sich eine erhebliche Steigerung der Lebensqualität der Patienten[22][11][18][19] u. a. durch eine Verbesserung der Fatigue-Symptomatik.[18][19] Eculizumab wurde gut vertragen und das Nebenwirkungsprofil war vergleichbar mit der der Placebogruppe.[19] Da Eculizumab einen Teil der Komplementkaskade blockiert und damit auch die Abwehr von Bakterien behindert, ist das Risiko einer Meningokokkeninfektion mit der Folge einer Hirnhautentzündung erhöht. Aus diesem Grund müssen alle Patienten mindestens zwei Wochen vor Therapiebeginn gegen Meningokokken geimpft werden.[22][11][23] Langzeitüberlebensdaten von Patienten, die seit bis zu 8 Jahren mit Eculizumab behandelt werden, zeigen, dass sich die Lebenserwartung der Patienten an die der normalen Bevölkerung angleicht.[8]
Im Dezember 2018 erteilte die amerikanische Zulassungsbehörde FDA eine Zulassung für Ravulizumab (Handelsname: Ultomiris), das ebenfalls ein monoklonaler Antikörper gegen Komplementfaktor C5 ist.[24] In der EU wurde Ravulizumab am 2. Juli 2019 zugelassen.[25]
Die deutsche Leitlinie empfiehlt Eculizumab und Ravulizumab gleichermaßen zur Therapie symptomatischer Patienten.[3]
Forschungsgeschichte
Der amerikanische Hämatologe Charles Parker aus Salt Lake City, Utah hat sich intensiv mit der Forschungsgeschichte der paroxysmalen nächtlichen Hämoglobinurie auseinandergesetzt. In einem umfangreichen Artikel von 2002 teilt er die Forschungsgeschichte in eine „frühe Geschichte“ und eine „moderne Geschichte“ ein. Die erste Phase war durch die Beschreibung und Definition des Krankheitsbildes geprägt. Die Entdeckung des alternativen Aktivierungsweges des Komplementsystems markierte einen Wendepunkt, weil von nun an die molekularbiologischen Grundlagen der Krankheit aufgedeckt wurden. Ihren vorläufigen Höhepunkt erreichte die Forschung 1993 mit der Aufdeckung der genetischen Ursache. Parker fragt, warum eine derartig seltene Erkrankung verhältnismäßig viel Aufmerksamkeit durch die Forschung erhielt. Er glaube, die „elegante Komplexität“ der Natur, die in der Pathophysiologie der PNH zu erkennen sei, übe eine Faszination auf Hämatologen aus. Die PNH-Forschung trieb einerseits den Erkenntnisfortschritt in der Erforschung des Immunsystems voran. Andererseits sei sie ein Beispiel dafür, wie beiläufige Beobachtungen aus benachbarten Forschungsgebieten entscheidende Erkenntnisse füreinander bergen können.[9]
Die Beschreibung der Krankheit
Die erste Beschreibung eines Patienten mit paroxysmaler nächtlicher Hämoglobinurie wird dem Greifswalder Arzt Paul Strübing zugerechnet, der 1882[26] noch als Assistenzarzt den Fall des 29-Jährigen Stellmachermeisters Carl G. beschreibt. Dieser bemerkte seit 1876 eine morgendliche dunkle Verfärbung des Urins, während seine Haut zusehends blasser wurde und sich grau-gelblich färbte. Die Symptomatik war anfallsartig verstärkt und ging mit Abgeschlagenheit, Schwindel und Herzklopfen sowie mit Schmerzen in der Milz- und Nierengegend einher. Strübing fand heraus, dass der Urin Hämoglobin beinhaltete, aber keine roten Blutkörperchen. Ebenso beobachtete er, dass sich nach schweren Anfällen auch das Blutplasma rot färbte. Hieraus zog er den (richtigen) Schluss, dass eine Zerstörung roter Blutkörperchen in den Blutgefäßen vorliegen müsse. Er nahm an, dass der Schlaf eine entscheidende Rolle spiele, weil zwar nach dem nächtlichen Wecken des Patienten dunkler Urin ausgeschieden wurde, sonst aber nicht. Strübing war bekannt, dass Erythrozyten in einem sauren Milieu (unter Inkubation mit CO2, denn eine höhere CO2-Konzentration im Blut säuert dieses an) schneller lysieren. Er stellte daher die Hypothese auf, dass die Ursache der Hämolyse darin liege, dass sich im Schlaf durch langsameren Blutfluss die CO2-Konzentration erhöhe. Dadurch werde das Blut saurer, was die Hämolyse verursache. Strübing versuchte, seine Hypothese zu untermauern: Er verabreichte seinem Patienten Säure, um einen Anfall auszulösen, was jedoch nicht gelang.[9]
Tatsächlich ist Strübings Beschreibung nicht die älteste Veröffentlichung, die einen Fall von PNH beschreibt. Eine ältere Beschreibung stammt beispielsweise von dem britischen Arzt William Gull (1866), die älteste könnte aus dem Jahr 1793 sein. Strübings Verdienst war es aber, die PNH als eigenständige Krankheit zu erkennen, und sie von Hämoglobinurien durch Kälte (Autoimmunhämolytische Anämie vom Kältetyp) sowie der Marschhämoglobinurie abzugrenzen, weshalb er als Erstbeschreiber anerkannt wird. Strübing schlug später eine Karriere außerhalb der Hämatologie ein, weswegen er die Krankheit nicht weiter verfolgte.[9]
Ab 1908 wurde die Krankheit vermehrt beforscht und beschrieben, ein Übersichtsartikel von 1949[27] nennt 73 Fallbeschreibungen, die zwischen 1908 und 1949 veröffentlicht wurden. Aus diesem Zeitraum sind die Arbeiten der Italiener Ettore Marchiafava und Ferdinando Micheli, die 1928 bzw. 1931 Fälle aufarbeiteten und als Syndrom beschrieben, hervorzuheben. Aufgrund ihrer Leistungen wurde die Erkrankung bis in die 1960er Jahre hinein als Marchiafava-Micheli-Syndrom bezeichnet, ehe der Begriff aus der Mode kam. Der Name „paroxysmale nächtliche Hämoglobinurie“ wurde 1928 von dem Amsterdamer Arzt J. Enneking erstmals verwendet und setzte sich zusehends durch.[9]
Van den Bergs Experimente
Eine wichtige Arbeit für das Verständnis der Krankheit wurde von dem Niederländer Hijmans van den Berg geleistet. Dieser zeigte 1911, dass die Erythrozyten von PNH-Patienten im Serum in vitro (also im Reagenzglas) in einer CO2-Atmosphäre lysieren – und zwar im Serum des Patienten, aber auch wenn sie in die Blutseren blutgruppengleicher Gesunder gegeben wurden. Gleichzeitig zeigte er, dass die Erythrozyten gesunder Versuchspersonen die gleiche Behandlung unbeschadet überstehen. Damit war bewiesen, dass die Ursache der Hämolyse in den Erythrozyten lag, und dass es sich nicht um einen krankhaften Faktor im Blutserum handeln konnte.[9]
Van den Bergs Experimente bargen noch eine weitere wichtige Erkenntnis, weil sie erstmals das Komplementsystem ins Spiel brachten. Zu dieser Zeit waren drei Komponenten der humoralen Immunantwort bekannt: Antigen, Antikörper und das Komplementsystem, das die Funktion der Antikörper ergänzt (komplementiert). Über das Komplementsystem war bekannt, dass es durch Hitze zerstört und seine Funktion durch Gabe einer geringen Menge Blutserum (das Komplementfaktoren enthält) wiederhergestellt werden kann. Van den Berg beobachtete, dass tatsächlich keine Hämolyse stattfand, wenn das Serum zuvor erhitzt wurde. Das deutete darauf hin, dass Komplement an der Hämolyse beteiligt ist. Aber, anders als es vor dem Wissensstand der Zeit zu erwarten war, konnte die Hämolyseaktivität nicht durch Zugabe von frischem Serum wiederhergestellt werden. Van den Berg verwarf daher die Annahme, dass das Komplementsystem an der Hämolyse beteiligt sein könnte.[9]
Heute wissen wir, dass die damals bekannten Eigenschaften des Komplementsystems auf den klassischen Weg der Komplementaktivierung zutreffen. Dieser ist die Komplementaktivierung durch Bindung eines Antikörpers an ein Antigen. Dieser Mechanismus spielt bei der autoimmunhämolytischen Anämie eine Rolle, nicht aber bei der PNH. Bei der PNH binden Komplementfaktoren direkt an Strukturen auf der Zelloberfläche und aktivieren dadurch den Membranangriffskomplex – der alternative Weg der Aktivierung, dessen Existenz erst in den 1960er Jahren bewiesen wurde. Die klassische Aktivierung über die Bildung von Antikörper-Antigen-Komplexen funktioniert auch noch bei hohen Verdünnungen, worauf die Wiederherstellung der Komplementaktivität durch Zugabe geringer Mengen Serum beruht. Die alternative Aktivierung ist dagegen schon bei geringer Verdünnung der Komplementfaktoren nicht mehr möglich. Deswegen konnte van den Berg zwar die Hämolyse durch Erhitzung des Komplements verhindern, bei der Zugabe der frischen Komplementfaktoren waren diese aber zu stark verdünnt, um über den alternativen Weg aktiviert werden zu können.[9]
Thomas Hale Ham
Ab 1937 begann Thomas Hale Ham, ein Arzt aus Cleveland, Ohio, sich mit der PNH zu beschäftigen. Seine Arbeiten sollten richtungsweisend für die Forschung der nächsten Jahrzehnte werden. Wie Strübing, dessen Publikation Ham nicht kannte, beschäftigte er sich zunächst mit dem Zusammenhang zwischen Schlaf und Hämoglobinurie. Er konnte durch Verschieben des Schlafrhythmus eines Studienpatienten zeigen, dass die Hämoglobinurie vom Schlaf, und nicht von der Tageszeit, abhängt. Ferner konnte er bei Studienpatienten hämolytische Anfälle auslösen, indem er ihnen Säure gab, und umgekehrt Anfälle verhindern, wenn er ihr Blut alkalisierte (durch Gabe von Basen oder indem er sie im Schlaf mit einer eisernen Lunge hyperventilieren ließ). Wie auch Strübing glaubte Ham, die Ursache der Hämolyse liege in einer erhöhten CO2-Konzentration im Blut durch eine Hypoventilation im Schlaf.[9]
Als nächstes ergänzte Ham seine Experimente am Menschen durch Laborexperimente. Er wiederholte van den Bergs wesentliche Experimente, wobei er allerdings nicht auf van den Bergs Arbeiten verweist – es ist nicht klar, ob sie ihm zu diesem Zeitpunkt noch unbekannt waren. Auch Ham schlussfolgerte richtig, dass die Ursache der Krankheit in den Erythrozyten des Erkrankten liegt, während der hämolysierende Faktor ein normaler Bestandteil des Blutes sein muss. Ham begründete, warum am Prozess der Hämolyse keine Antikörper beteiligt sind, kam dadurch aber zu dem Fehlschluss, dass ein bislang unbekanntes, vom Komplementsystem verschiedenes System die Hämolyse vermitteln müsse.[9]
Aus der in dieser Experimentenreihe gemachten Beobachtung, dass PNH-Erythrozyten bei Ansäuerung des Blutes lysieren, wurde später ein diagnostischer Test entwickelt, der Ham-Test. Dieser war für 50 Jahre die Standardmethode in der Diagnostik, bis er durch die Durchflusszytometrie ersetzt wurde.[28]
1939[29] veröffentlichte Ham mit seinem Kollegen John Holmes Dingle eines der einflussreichsten Paper in der Forschungsgeschichte der PNH, in dem sie detailliert darlegen, dass ein bislang unbekannter Mechanismus die Erythrozyten lysiert. Der Befund, dass keine Antikörper daran beteiligt sind, war völlig neu und passte nicht in das bisherige Bild der humoralen Immunantwort. Obwohl sie mehrere deutliche Hinweise auf eine Rolle des Komplementsystems beschreiben, und vermutlich auch selbst daran glaubten, dass dieses für die Hämolyse verantwortlich ist, behaupten sie im Diskussionsteil des Artikels, das Komplementsystem sei nicht der lysierende Faktor. Vermutlich waren sie in ihren Schlussfolgerungen zurückhaltend, weil sie ahnten, dass ihre Ergebnisse in der Fachwelt auf Skepsis und Ablehnung stoßen würden. Ein im Rückblick bedeutsamer Nebenbefund war die Beobachtung, dass bei ihrem Testpatienten nur 15 % der roten Blutkörperchen von der Hämolyse betroffen waren.[9]
Entdeckung des alternativen Wegs der Komplementaktivierung
Das Jahrzehnt nach Hams und Dingles Publikation sah kaum Fortschritte. Die Forschung sammelte weiter Daten, die für und gegen eine Rolle des Komplementsystems sprachen. Eine – im Rückblick – bedeutsame Entdeckung war, dass der hämolytische Prozess bei der PNH (über die damals unbekannte alternative Komplementaktivierung) von Magnesium-Ionen (Mg2+) abhängt. Darin unterscheidet er sich von der autoimmunhämolytischen Anämie.[9]
Eine erste Erklärung, welcher Teil des Immunsystems die Hämolyse bei der PNH vermittelt, lieferte Louis Pillemer. Pillemer isolierte 1954 bei dem Versuch, den damals so bezeichneten dritten Komplementfaktor zu isolieren (der eigentlich aus mehreren Komplementfaktoren besteht), zufällig Properdin – ein Protein, das die Anlagerung des Komplementfaktors C3b an Faktor B unterstützt. Pillemer bemerkte, dass Properdin nur in Anwesenheit von Komplement und Magnesium aktiv war. Dabei wirkte es an der Zerstörung von Bakterien, Viren und „bestimmter roter Blutkörperchen“ mit – PNH-Erythrozyten. Dabei fiel auf, dass Antikörper für die Properdinaktivität nicht benötigt wurden. Zwei Jahre später wies Pillemer mit seinen Kollegen Carl F. Hinz und William S. Jordan nach, dass Properdin zwar für die Hämolyse von PNH-Erythrozyten zwingend notwendig ist, für die Hämolyse bei der autoimmunhämolytischen Anämie jedoch verzichtbar. Pillemers Studien an Properdin und der PNH lieferten starke Hinweise darauf, dass es eine Antikörper-unabhängige Komplementaktivierung gibt. Allerdings konnte sich Pillemers Hypothese vom Properdin-System unter Komplementsystem-Forschern nicht durchsetzen, insbesondere deswegen, weil sich technisch bedingt kleine Mengen Antikörper in seinen Proben fanden. Seine Kollegen nahmen daher an, dass diese die Komplementaktivierung erklärten. Pillemer starb 1957 nach einem Treffen mit anderen auf dem Gebiet tätigen Wissenschaftlern an einer Überdosis Schlafmitteln.[9] Unter Hämatologen hingegen fand das Properdin-System als Erklärung für die Hämolyse der PNH schnell weitgehende Akzeptanz, da es am besten zu den Beobachtungen der vorangegangenen Jahrzehnte passte. Erst nach der Entwicklung besserer Aufbereitungsmethoden und 10 Jahre nach Pillemers Tod konnten seine Vermutungen bestätigt werden. Bis 1980 waren die Bestandteile des alternativen Aktivierungsweges aufgeklärt.[9]
PNH ist ein Mosaik
Nach Ham und Dingle beobachteten Forscher immer wieder, dass niemals alle Erythrozyten von PNH-Patienten von Hämolyse betroffen waren, was nahelegte, dass sich zwei unterschiedliche Populationen von Erythrozyten im Blut der Betroffenen finden (also ein Mosaik bilden). Die britischen Hämatologen Wendell F. Rosse und John Dacie untersuchten diese Frage 1966 genauer. Durch genaue Beobachtungen des Verlaufs von Hämolysen in gesundem Blut und dem Blut von PNH-Patienten wiesen sie nach, dass PNH-Kranke einen gewissen Anteil hämolyseanfälliger Erythrozyten haben, während der Rest ihrer roten Blutkörperchen eine normale Empfindlichkeit gegenüber dem Komplementsystem zeigt. Dabei lag der Anteil hämolyseanfälliger Erythrozyten zwischen 4 und 80 %, was die unterschiedliche Ausprägung der Symptome erklärte. Ursprünglich ging man davon aus, dass es nur zwei Populationen von PNH-Zellen gebe: eine, die sehr anfällig gegenüber der Hämolyse durch das Komplementsystem ist, und eine, die nicht anfällig ist. Durch weitere Studien in den 1970ern musste diese Einschätzung jedoch korrigiert werden, denn bei manchen Patienten ließen sich mehrere Erythrozytenpopulationen nachweisen, deren Hämolyse-Anfälligkeit zwischen den beiden ursprünglich postulierten Gruppen lag.[9]
Ebenfalls 1969 konnte gezeigt werden, dass auch die Blutplättchen und bestimmte Immunzellen von PNH-Patienten anfälliger für die komplementvermittelte Hämolyse sind, was die These stützte, dass es einen gemeinsamen Vorgänger dieser Zellen gibt, und dass die Ursache der PNH eine somatische Mutation ist. Zu diesem Zeitpunkt war die Existenz blutbildender Stammzellen, aus denen alle Blutzelllienen hervorgehen können, noch nicht bewiesen und hoch umstritten. Die Ergebnisse der PNH-Forschung lieferten hier Argumente für die Befürworter dieser Theorie.[9]
Entdeckung der Komplementregulatoren
Edward M. Hoffmann, ein Hämatologe aus Florida, berichtete 1969 als erster von einem Faktor in Erythrozyten, der die komplement-vermittelte Hämolyse unterbinden kann. Diesen nannte er decay accelerating factor (DAF), weil er den Abfall der Komplementaktivität beschleunigt. DAF (heute als CD55 bezeichnet) verhindert die Umwandlung des Komplementfaktors C3 zu C3b. Damit blockiert er sowohl die Komplementaktivierung über den klassischen als auch über den alternativen Weg. 1983 berichteten Anne Nicholson-Weller und Kollegen, dass PNH-Erythrozyten DAF fehle. Dass dieser Mangel an DAF eine ursächliche Rolle in der Pathogenese der Erkrankung spielen werde, war schnell allgemein akzeptiert. Er erklärte allerdings nicht alles, denn es gab auch Hinweise auf eine gestörte Regulation des Membranangriffs-Komplexes (MAC). Der Regulator für MAC konnte erst 1989 identifiziert werden. Seine Entdecker gaben ihm den Namen membrane inhibitor of reactive lysis (MIRL, zu Deutsch „Membran-Inhibitor der reaktiven Lyse“), heute läuft er unter dem Kürzel CD59.[9]
Mit der Kenntnis dieser beiden Regulatoren konnte erklärt werden, warum PNH-Erythrozyten unterschiedlich anfällig für die Lyse sind. Erythrozyten, denen beide Regulatoren vollständig fehlen, sind am anfälligsten für das Komplementsystem. Es gibt aber Erythrozyten, deren Anfälligkeit geringer ausgeprägt ist, weil sie noch in geringerem Maße CD55 und CD59 auf ihrer Oberfläche haben. Während die Menge von CD59 noch ausreicht, um den MAC zu kontrollieren, kann CD55 die Umwandlung von C3 zu C3b nicht mehr verhindern.[9]
Der GPI-Anker
Zwischen die Entdeckung von DAF und MIRL fällt die Entdeckung des GPI-Ankers. Hierzu, und zur Erkenntnis der Bedeutung für die PNH, führten einige zufällige Beobachtungen (Serendipität). Die erste stammt von 1951, als Forscher, die sich eigentlich für Leukämien interessierten, zufällig auch Leukozyten eines PNH-Patienten untersuchten und bemerkten, dass auf diesen die alkalische Phosphatase fehlte. Andere Forscher berichteten in den 1950ern, dass auf PNH-Erythrozyten auch keine Acetylcholinesterase und keine 5′-Nukleotidase zu finden sind. Diese Enzyme stehen in keinem ursächlichen Zusammenhang mit der Erkrankung, warfen aber die Frage auf, warum drei nicht miteinander in Verbindung stehende Enzyme auf PNH-Zellen fehlen. Die Antwort kam 1985, nachdem ein Science-Artikel über Experimente mit einem bakteriellen Enzym berichtet hatte, die in den 1970ern durchgeführt worden waren. Dieses Enzym war eine Phospholipase, die Phosphatidylinsositole spaltet. Der Artikel berichtet, dass der Einsatz der Phospholipase zur Freisetzung von Alkalischer Phosphatase, Acetylcholinesterase und 5’-Nucleotidase, sowie von Thy1 (CD90), von Zellmembranen führt. Wissenschaftler der Universität von New York erkannten die Verbindung zur PNH. Sie besorgten sich die Phospholipase und konnten zeigen, dass auch DAF (CD55) durch diese von der Zellmembran gelöst wird. Für die Forscher war damit klar, dass jedes Protein, dass auf der Oberfläche von PNH-Erythrozyten fehlt, den gleichen Anker hat, und das andersherum jedes Protein, das normalerweise über diesen Anker mit der Zellmembran verbunden ist, auf PNH-Zellen fehlen muss.[9]
Entschlüsselung der genetischen Ursache
Bis 1987 war die Struktur dieses Ankers, Glycosylphosphatidylinositol, aufgeklärt. Man nahm bereits seit den 1960ern an, dass die Ursache der PNH in einer somatischen Mutation liegen könne. Als der Defekt des GPI-Ankers als zentral in der Entstehung der PNH bekannt wurde, stand die Forschung vor dem Problem, den ursächlichen Gendefekt zu identifizieren. Theoretisch waren Schädigungen in jedem der zahllosen Gene denkbar, die an der GPI-Synthese und der Verankerung der Proteine in der Zellmembran beteiligt sind. Erste Experimente deuteten auch darauf hin, dass die Störung in vielen unterschiedlichen Stellen liegen könne.[9]
Zu Hilfe kam – wieder einmal – ein glücklicher Zufall. In den 1970ern wurde eine Linie von Lymphomzellen herangezüchtet, um die genetische Basis der Krebsentstehung zu erforschen. Zufälligerweise fehlte diesen Zellen auch CD90, was im Kontext der Lymphomforschung nebensächlich war. Mit dem Wissen um die Verankerung von CD90 durch GPI nutzten japanische Forscher um Taroh Kinoshita diese gut erforschte Zellreihe, um 1993 die genetische Ursache der PNH zu entschlüsseln.[9]
Kinoshita und seine Kollegen wiesen 1993 in einer Reihe anspruchsvoller Experimente[30][31][32] nach, dass der genetische Defekt, der in den Lymphomzellen zum Fehlen des GPI-Ankers führt, der gleiche sein muss, der in den von ihnen untersuchten PNH-Zellen vorlag. Dies schlossen sie daraus, dass die Zellen ohne GPI-Anker kein N-Acetylglucosaminphosphatidylinositol herstellen konnten – was der erste Schritt der GPI-Synthese ist. Sie konnten ein Gen identifizieren, das für diesen Schritt verantwortlich erschien. Sie vervielfältigten dieses aus einer gesunden Zelle und übertrugen es auf GPI-lose Lymphomzellen. Nach der Übertragung synthetisierten die Zellen N-Acetylglucosaminphosphatidylinositol. Die Forscher tauften das Gen auf den Namen PIGA (Phosphatidylinosytol Glycan Class A). Sie verglichen den Code des Gens GPI-tragender und GPI-loser Zellen und fanden in den GPI-losen Zellen Unterschiede. Die Wissenschaftler bewiesen, dass es sich um erworbene Mutationen handelt, indem sie gesunde und kranke Zellen des gleichen Patienten verglichen. Die PIGA-Mutation fand sich nur in den PNH-Zellen. Wäre die PIGA-Mutation eine Keimbahnmutation, wäre sie in jeder Zelle nachzuweisen. Einen starken Hinweis darauf, dass diese Mutation in einer blutbildenden Stammzelle auftritt, lieferte die Arbeitsgruppe mit dem Nachweis der gleichen Mutation in B-Lymphozyten und Neutrophilen Granulozyten des gleichen Patienten. Schlussendlich konnte sie PIGA auf dem X-Chromosom lokalisieren.[9]
Bislang wurden rund 150 verschiedene Mutationen in PIGA beschrieben, deren Auswirkungen auf die GPI-Synthese von einer Funktionseinschränkung bis zum kompletten Verlust reichen. Diese Vielfalt des Genotyps von PNH-Stammzellen erklärt, warum sich PNH-Zellen in ihrer Anfälligkeit für die Hämolyse unterscheiden.[33]
Weblinks und Literatur
- Nature Videos: PNH: When blood machinery goes wrong – Laienverständliches Video zur Entstehung der PNH. Englische Originalsprache, für deutsche Untertitel zunächst im Videofenster Untertitel einschalten, dann bei Optionen -> Untertitel "Automatisch übersetzen" wählen.
- Stiftung lichterzellen – Stiftung zur Förderung der Forschung sowie zur Unterstützung Erkrankter.
- Aplastische Anämie & PNH e.V.
- Aktuelle Leitlinie zur Paroxysmalen nächtlichen Hämoglobinurie auf onkopedia.com
- A Roth, U. Duhrsen: Paroxysmale nächtliche Hämoglobinurie In: Deutsches Ärzteblatt, 2007, 104, S. 192–197.
- Anita Hill, Amy E. DeZern, Taroh Kinoshita, Robert A. Brodsky: Paroxysmal nocturnal haemoglobinuria. In: Nature Reviews Disease Primers. Band 3, Nr. 1, 18. Mai 2017, ISSN 2056-676X, S. 1–14, doi:10.1038/nrdp.2017.28.
- Charles J. Parker: Historical aspects of paroxysmal nocturnal haemoglobinuria: 'defining the disease'. In: British Journal of Haematology. Band 117, Nr. 1, April 2002, ISSN 0007-1048, S. 3–22, doi:10.1046/j.1365-2141.2002.03374.x.
Einzelnachweise
- A Roth, U. Duhrsen: Paroxysmale nächtliche Hämoglobinurie In: Dtsch Arztebl, 2007, 104, S. 192–197
- G. Socié, H. Schrezenmeier, P. Muus, I. Lisukov, A. Röth: Changing prognosis in paroxysmal nocturnal haemoglobinuria disease subcategories: an analysis of the International PNH Registry. In: Internal Medicine Journal. Band 46, Nr. 9, 2016, ISSN 1445-5994, S. 1044–1053, doi:10.1111/imj.13160.
- Jörg Schubert, Alexander Röth, Peter Bettelheim, Georg Stüssi, Britta Höchsmann, Jens Panse, Tim Henrik Brümmendorf, Hubert Schrezenmeier: Paroxysmale nächtliche Hämoglobinurie (PNH). DGHO-Leitlinie auf onkopedia.com, Stand November 2019. Zuletzt abgerufen am 20. März 2020.
- Anita Hill, Amy E. DeZern, Taroh Kinoshita, Robert A. Brodsky: Paroxysmal nocturnal haemoglobinuria. In: Nature Reviews Disease Primers. Band 3, Nr. 1, 18. Mai 2017, ISSN 2056-676X, S. 1–14, doi:10.1038/nrdp.2017.28.
- Lucio Luzzatto: Hämolytische Anämien und Anämien durch akuten Blutverlust, für die deutsche Ausgabe von Andreas Loew und Julia Jesse. In: Harrisons Innere Medizin, 19. Auflage. ABW-Verlag, Berlin 2016. ISBN 978-3-88624-560-4. S. 804
- P Hillmen, SM Lewis, M Bessler, L Luzzatto, JV. Dacie: Natural history of paroxysmal nocturnal hemoglobinuria. (PDF) In: The New England Journal of Medicine 1995, 333, S. 1253–1258.
- RP de Latour, JY Mary, C Salanoubat et al.: Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. In: Blood, 2008, 112, S. 3099–3106, PMID 18535202
- RJ Kelly, A Hill, LM Arnold et al.: Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. In: Blood, 2011, 117, S. 6786–6792, PMID 21460245
- Charles J. Parker: Historical aspects of paroxysmal nocturnal haemoglobinuria: 'defining the disease'. In: British Journal of Haematology. Band 117, Nr. 1, April 2002, ISSN 0007-1048, S. 3–22, doi:10.1046/j.1365-2141.2002.03374.x.
- Neal S. Young: Aplastic Anemia. In: New England Journal of Medicine. Band 379, Nr. 17, 25. Oktober 2018, ISSN 0028-4793, S. 1643–1656, doi:10.1056/NEJMra1413485, PMID 30354958, PMC 6467577 (freier Volltext).
- H. Schrezenmeier, B. Hochsmann: Eculizumab opens a new era of treatment for paroxysmal nocturnal hemoglobinuria. In: Expert review of hematology, 2009, 2, S. 7–16, PMID 21082989
- C Parker, M Omine, S Richards et al.: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. In: Blood, 2005, 106, S. 3699–3709, PMID 16051736
- Ham's Test (Acidified Serum Test). Abgerufen am 24. Juli 2020.
- What is the role of the sucrose lysis test in the workup of paroxysmal nocturnal hemoglobinuria (PNH)? Abgerufen am 24. Juli 2020.
- C. Parker: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. In: Blood. Band 106, Nr. 12, 1. Dezember 2005, ISSN 0006-4971, S. 3699–3709, doi:10.1182/blood-2005-04-1717, PMID 16051736, PMC 1895106 (freier Volltext) – (bloodjournal.org [abgerufen am 24. Juli 2020]).
- RA Brodsky, GL Mukhina, S Li et al.: Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. In: American Journal of Clinical Pathology, 2000, 114, S. 459–466, PMID 10989647. PMC 4124633 (freier Volltext).
- Thomas C. Thomas, Scott A. Rollins, Russell P. Rother, Michelle A. Giannoni, Sandra L. Hartman: Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. In: Molecular Immunology. Band 33, Nr. 17, 1. Dezember 1996, ISSN 0161-5890, S. 1389–1401, doi:10.1016/S0161-5890(96)00078-8 (sciencedirect.com [abgerufen am 29. März 2020]).
- RA Brodsky, NS Young, E Antonioli et al.: Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. In: Blood, 2008, 111(4), S. 1840–1847, PMID 18055865
- P Hillmen, NS Young, J Schubert et al.: The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. In: The New England journal of medicine, 2006, 355, S. 1233–1243, PMID 16990386. doi:10.1056/NEJMoa061648.
- P Hillmen, C Hall, JC Marsh et al.: Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. In: The New England journal of medicine, 2004, 350, S. 552–559. doi:10.1056/NEJMoa031688.
- P Hillmen, P Muus, U Duhrsen et al.: Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. In: Blood, 2007, 110, S. 4123–4128, PMID 17702897
- A Roth, U Duhrsen, H Schrezenmeier, J. Schubert: Paroxysmal nocturnal hemoglobinuria (PNH). Pathogenesis, diagnosis and treatment. In: Dtsch med Wochenschr, 2009, 134(9), S. 404–409, doi:10.1055/s-0028-1124013
- Alexion: Fachinformation Soliris (Eculizumab) (PDF; 358 kB), 2012.
- FDA approves new treatment for adult patients with rare, life-threatening blood disease, PM FDA vom 21. Dezember 2018, abgerufen am 2. Januar 2019
- ULTOMIRIS® (ravulizumab) Receives Marketing Authorization from European Commission for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH) - Pressemitteilung von Alexion vom 3. Juli 2019. Zuletzt abgerufen am 21. März 2020.
- P. Strübing: Paroxysmale Haemoglobinurie. In: DMW - Deutsche Medizinische Wochenschrift. Band 8, Nr. 01, Januar 1882, ISSN 0012-0472, S. 1–3, doi:10.1055/s-0029-1196307.
- John Marks: THE MARCHIAFAVA MICHELI SYNDROME (Paroxysmal Nocturnal Haemoglobinuria). In: QJM: An International Journal of Medicine. Band 18, Nr. 2, 1. April 1949, ISSN 1460-2725, S. 105–121, doi:10.1093/oxfordjournals.qjmed.a066527.
- Jeffrey J. Pu, Robert A. Brodsky: Paroxysmal Nocturnal Hemoglobinuria from Bench to Bedside. In: Clinical and Translational Science. Band 4, Nr. 3, Juni 2011, S. 219–224, doi:10.1111/j.1752-8062.2011.00262.x, PMID 21707954, PMC 3128433 (freier Volltext).
- Thomas Hale Ham, John H. Dingle: STUDIES ON DESTRUCTION OF RED BLOOD CELLS. II. CHRONIC HEMOLYTIC ANEMIA WITH PAROXYSMAL NOCTURNAL HEMOGLOBINURIA: CERTAIN IMMUNOLOGICAL ASPECTS OF THE HEMOLYTIC MECHANISM WITH SPECIAL REFERENCE TO SERUM COMPLEMENT 1. In: Journal of Clinical Investigation. Band 18, Nr. 6, 1. November 1939, ISSN 0021-9738, S. 657–672, doi:10.1172/JCI101081, PMID 16694699, PMC 434913 (freier Volltext).
- M. Takahashi, J. Takeda, S. Hirose, R. Hyman, N. Inoue: Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. In: Journal of Experimental Medicine. Band 177, Nr. 2, 1. Februar 1993, ISSN 0022-1007, S. 517–521, doi:10.1084/jem.177.2.517, PMID 8426120, PMC 2190897 (freier Volltext) – (rupress.org [abgerufen am 29. März 2020]).
- T Miyata, J Takeda, Y Iida, N Yamada, N Inoue: The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. In: Science. Band 259, Nr. 5099, 26. Februar 1993, ISSN 0036-8075, S. 1318–1320, doi:10.1126/science.7680492.
- Junji Takeda, Toshio Miyata, Kazuyoshi Kawagoe, Yoshiyasu Iida, Yuichi Endo: Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. In: Cell. Band 73, Nr. 4, 21. Mai 1993, ISSN 0092-8674, S. 703–711, doi:10.1016/0092-8674(93)90250-T, PMID 8500164.
- Charles J. Parker: Paroxysmal Nocturnal Hemoglobinuria: An Historical Overview. In: Hematology. Band 2008, Nr. 1, 1. Januar 2008, ISSN 1520-4391, S. 93–103, doi:10.1182/asheducation-2008.1.93 (ashpublications.org [abgerufen am 29. März 2020]).