Pseudorotation
Pseudorotation ist ein Phänomen, das in der Stereochemie auftritt und den schnellen Wechsel der Positionen von Atomen oder atomaren Baugruppen in Molekülen und Komplexverbindungen beschreibt. Durch die Pseudorotation entstehen kurzzeitig Konformationsisomere einer Verbindung, das heißt die räumliche Anordnung der Atome zueinander ändert sich, ohne dass chemische Bindungen zwischen den Atomen gelöst und neu gebildet werden. Pseudorotationen erfolgen so schnell, dass Messgeräte wie NMR-Spektrometer den Wechsel der Atompositionen nicht erfassen und nur eine zeitlich gemittelte Struktur der Verbindung wiedergeben können. Die Pseudorotation zählt zu den sogenannten intramolekularen Vorgängen.
Der Begriff wurde im Jahr 1947 von den US-amerikanischen Physikochemikern John E. Kilpatrick, Kenneth S. Pitzer und Ralph Spitzer geprägt, die eine Untersuchung über die Thermodynamik und Molekülstruktur des Kohlenwasserstoffs Cyclopentan vorlegten.[1]
Rotation und Pseudorotation
Vor der Entwicklung quantenmechanischer Theorien zur Chemischen Bindung und Molekülstruktur wurde der dreidimensionale Bau von organischen Molekülen durch Modelle anschaulich gemacht. Einfachbindungen wurden durch Stäbchen dargestellt, die Atome durch Kugeln, welche mit den Stäbchen verbunden wurden. (Heutige Modelle verwenden an Stelle der Kugeln im Fall von Kohlenstoffatomen Metall- oder Plastik-Bausteine, welche die idealen Valenzwinkel – 109° 28‘ (tetraedrisch), 120° (trigonal), 180° (diagonal) – vorgeben.)
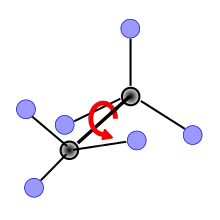
Die Kugel-Stab-Modelle konnten zu der Ansicht verführen, dass Teile eines Moleküls um C-C-Einfachbindungen „frei“ gedreht werden könnten („Freie Drehbarkeit“). Das einfachste Beispiel war der Kohlenwasserstoff Ethan: Die beiden Methylgruppen lassen sich im Modell gegeneinander drehen.
Der Begriff der Drehung (Rotation) ist zwar anschaulich, weil er auf einem einfachen Modell basiert, aber mathematisch-physikalisch gesehen ändern sich durch Schwingungen die internen Koordinaten der Atome, aus denen das Molekül aufgebaut ist.
Es zeigte sich jedoch, dass für die Rotation im Ethan-Molekül Energie aufgewendet werden muss, und eine (Energie-)Barriere zu überwinden ist. Verglichen mit der Bindungsenergie der C-C-Bindung ist die für die Rotation nötige Energie sehr klein.
Bei kleineren Cycloalkanen ist – wie man an Kugel-Stab-Modellen erkennt – eine Rotation um C-C-Einfachbindungen nicht möglich, ohne diese Bindung aufzubrechen, d. h. mindestens den Betrag der Bindungsenergie zuzuführen. Aber Schwingungen, d. h. Veränderung der internen Atomkoordinaten sind noch möglich.
Die Studien am Cyclopentan führten Pitzer und Mitarbeiter zu der Auffassung, dass das Molekül "sich faltet" oder "kräuselt" (englisch: puckering). So kann eines der fünf C-Atome aus der gedachten Ebene eines regelmäßigen Fünfecks herausschwingen; durch das Molekül läuft eine Art Wellenbewegung. Diesen Vorgang nannten die Forscher „Pseudorotation“.
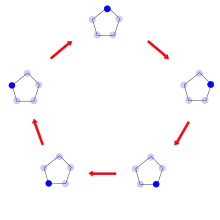
Ein weiterer Modus der Pseudorotation entsteht dadurch, dass sich zwei CH2-Gruppen relativ zu den anderen verdrehen (englisch: twist). Dabei zeigt der Atomverband zu jeder Zeit C2-Symmetrie, d. h. eine zweizählige Drehachse. Bildlich ist dieser Vorgang schwieriger darzustellen.
Pseudorotation zeigen auch die Moleküle von Cycloalkanen mit Ringgrößen >5, doch sind die Verhältnisse komplizierter und in einfachen Bildern nicht leicht zu zeigen.
Pitzer erkannte auch, worauf die Pseudorotation beim Cyclopentan und die Barriere der Rotation beim Ethan beruht: Die Natur vermeidet Drehwinkel (Torsionswinkel) von Null Grad, energetisch ungünstige sogenannte „ekliptische Wechselwirkungen“ benachbarter (vicinaler) C-H-Bindungen. Moleküle mit ekliptischen Wechselwirkungen von C-H- (und auch C-C-) Bindungen stehen unter "Torsionsspannung", welche nach dem Entdecker auch „Pitzer-Spannung“ genannt wird. Im Falle des Cyclopentans würden – wären die fünf C-Atome in einer Ebene angeordnet – alle benachbarten C-H-Bindungen einen Winkel von 0° bilden, also in ekliptischer Stellung sein. Dies wäre das Maximum der potentiellen Energie, durch Pseudorotation wird eine energetisch günstigere Anordnung erreicht. Die günstigste Anordnung sind zwei Konformationen, welche Spiegelsymmetrie (Cs) und C2-Symmetrie (zweizählige Drehachse) aufweisen: Briefumschlag- und Halbsessel-Form.
Zu Pseudorotationen weiterer cyclischer organischer Verbindungen siehe die Artikel über die einzelnen Kohlenwasserstoffe.
Acyclische Verbindungen aus der (Anorganischen) Chemie
Der bekannteste und energetisch günstigste Pseudorotations-Mechanismus wurde von dem amerikanischen Chemiker R. Stephen Berry (Berry-Pseudorotation) für Moleküle mit trigonal-bipyramidalem Aufbau am Beispiel von Phosphorpentafluorid PF5 beschrieben.[2]
Trigonal-bipyramidale Moleküle am Beispiel von PF5
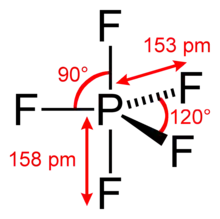
Die Berry-Pseudorotation findet in Molekülen mit trigonal-bipyramidalem Aufbau statt, das zentrale Atom weist dabei eine Koordinationszahl von fünf auf. Während bei Koordinationszahlen von vier (Tetraeder) oder sechs (Oktaeder) alle Atome den gleichen Abstand zum zentralen Teilchen einnehmen können, gibt es bei einer trigonalen Bipyramide zwei verschiedene Abstände. Drei der fünf Atome liegen dabei in der dreieckigen Grundfläche der Bipyramide (äquatoriale Position) und haben einen kürzeren Abstand zum Zentralatom als die zwei Atome an den Spitzen der Bipyramide (axiale oder apikale Position). In Molekülen der Form AX5, wie zum Beispiel das rechts dargestellte PF5, müssten sich daher drei Atome der Sorte X von den anderen zwei unterscheiden lassen, die Koordinationszahl fünf kann genauer als 3+2 beschrieben werden. Bereits 1953 entdeckte der amerikanische Chemiker Herbert S. Gutowsky bei Untersuchungen von PF5 mit Hilfe der 19F-NMR-Spektroskopie, dass anstatt der erwarteten zwei Signale für die Fluoratome nur ein einziges in den Spektren beobachtet werden kann.[3]
Mechanismus der Berry-Pseudorotation
In einem Molekül befinden sich die Atome nicht völlig starr an einer bestimmten Position, sondern führen in Abhängigkeit von der Umgebungstemperatur Schwingungen aus. Diese sogenannten Deformationsschwingungen führen zu periodischen Änderungen von Bindungslängen und Bindungswinkeln. Als Folge dieser Deformationsschwingungen bewegen sich bei der Berry-Pseudorotation die beiden Atome in apikaler Position (im Beispiel unten Nr. 4 und 5) aufeinander zu, der Bindungswinkel 4–Zentralatom–5 wird von 180° auf 120° verkleinert und die Bindungslängen verkürzen sich, während zwei äquatoriale Atome sich voneinander wegbewegen, der Bindungswinkel wird hier von 120° auf 180° aufgeweitet und der Abstand zum Zentralteilchen vergrößert sich (im Beispiel unten Nr. 1 und 2). Das verbleibende Atom in äquatorialer Position (im Beispiel Nr. 3) ist dabei der Angelpunkt (Fixpunkt), die anderen vier Atome bewegen sich relativ zu seiner Position. Während des Wechsels der Positionen wird ein Übergangszustand erreicht, bei dem die vier sich bewegenden Atome die Grundfläche einer quadratischen Pyramide bilden mit dem Angelpunkt als Pyramidenspitze. Am Ende der Pseudorotation folgt wieder der Grundzustand in Form einer trigonalen Bipyramide, wobei die ehemals apikalen Atome jetzt in äquatorialer Position zu finden sind und umgekehrt. Da jedes Atom in äquatorialer Position als Angelpunkt für die Pseudorotation dienen kann, besetzen nacheinander sämtliche Atome sowohl äquatoriale als auch apikale Positionen. Der Energiebedarf für den Übergang in den quadratisch-pyramidalen Zustand liegt bei der Berry-Pseudorotation für PF5 bei ca. 15 kJ/mol.[4]
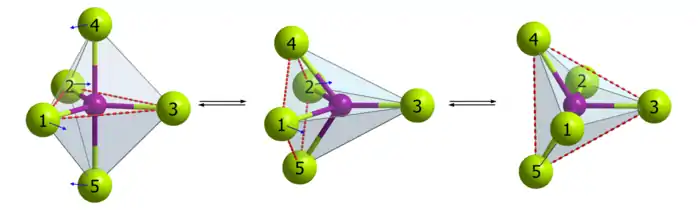
Da die Pseudorotation sehr schnell abläuft (Pseudorotationsfrequenzen bei Standardbedingungen ca. 105 s−1 für PF5 und ca. 102 s−1 für Phosphorpentachlorid PCl5), kann sie von einem NMR-Spektrometer nicht aufgelöst werden und lediglich eine zeitlich gemittelte Struktur mit nur einem Signal für alle fünf Atome kann in den Spektren beobachtet werden. Dadurch erscheinen die fünf Atome so, als wären sie gleichartig mit dem Zentralatom verbunden, obwohl sie in Wirklichkeit in einer schnell fluktuierenden trigonalen Bipyramide angeordnet sind.
Die Berry-Pseudorotation kann bei einigen Molekülen und Komplexen mit trigonal-bipyramidalem Aufbau beobachtet werden. Neben PF5 und PCl5 zählen hierzu auch Fe(PF3)5, Fe(CO)5 und SbMe5.
„Inverse Berry-Pseudorotation“
In Molekülen, die in ihrer Grundform als quadratische Pyramiden aufgebaut sind, kann eine Art „inverse Berry-Pseudorotation“ beobachtet werden. Bei Iodpentafluorid IF5 beispielsweise, pendelt ein Fluoratompaar in der Art einer sich öffnenden und schließenden Schere permanent vor und zurück und versetzt das gesamte Molekül in Schwingung. Wegen dieser durch die Pendelbewegung verursachten Schwingung bewegen sich dabei, im Gegensatz zur normalen Berry-Pseudorotation, alle Fluoratome mit, und es entsteht aus der quadratischen Pyramide kurzfristig eine trigonale Bipyramide. Bei dieser „inversen Berry-Pseudorotation“ sind der Grund- und Übergangszustand somit vertauscht. Allerdings beinhaltet diese Pseudorotation neben dem Berry-Mechanismus auch Bewegungsmuster der unten beschriebenen Lever- und Turnstile-Mechanismen und kann daher nicht als reine Umkehrung der Berry-Pseudorotation angesehen werden.
Weitere Pseudorotationsmechanismen
Neben der Berry-Pseudorotation existieren weitere Mechanismen, die mögliche Wechsel von Atompositionen in Molekülen erklären. Der Lever-Mechanismus (engl. lever = „Hebel“) beschreibt eine mögliche Pseudorotation von Fluoratomen in Schwefeltetrafluorid SF4 und Chlortrifluorid ClF3. Beide Moleküle haben unter Berücksichtigung von freien Elektronenpaaren ebenfalls einen trigonal-bipyramidalen Aufbau, wobei im Fall von SF4 eine äquatoriale Position durch das freie Elektronenpaar des Schwefels besetzt ist, in ClF3 nehmen die zwei freien Elektronenpaare des Chlors äquatoriale Positionen ein (siehe auch VSEPR-Modell). Die Pseudorotation wird dabei durch ein Fluoratom verursacht, das eine Bewegung ähnlich einem vor- und zurückgelegten Hebel ausführt, und dadurch die restlichen Fluoratome ebenfalls in Schwingung versetzt. Die Pseudorotation wird ausschließlich durch die Fluoratome ausgeführt, da freie Elektronenpaare, aufgrund ihres erhöhten Platzbedarfs, immer äquatoriale Positionen bevorzugen. Im Fall von SF4 kann auch eine Berry-Pseudorotation mit dem freien Elektronenpaar als Angelpunkt erfolgen. Als gemittelte Struktur ergibt sich für SF4 ohne Berücksichtigung des freien Elektronenpaars die Gestalt eines Disphenoids (ein gestrecktes Tetraeder). Der Energiebedarf für die Ausführung des Lever-Mechanismus liegt um ca. 42 kJ/mol höher als bei der Berry-Pseudorotation.[4] Bei ClF3 ist durch die zwei freien Elektronenpaare nur der Lever-Mechanismus möglich. Das T-förmige Molekül (ohne Berücksichtigung des freien Elektronenpaare) nimmt dabei als Übergangsform eine trigonal-planare Gestalt an.
Für oktaedrisch aufgebaute Moleküle existieren Berechnungen zum Energiebedarf des Wechsels von Chloratomen in Cis-trans-Isomeren der hypothetischen Verbindung Schwefeldichloridtetrafluorid SCl2F4. Der Wechsel der Chloratome von einer cis- in eine transständige Anordnung würde ca. 272 kJ/mol benötigen.[4] Die Bewegung würde von zwei Fluoratomen entgegen dem Uhrzeigersinn bei gleichzeitiger Rotation der anderen beiden im Uhrzeigersinn hervorgerufen werden. Diese Art der Pseudorotation wird als Turnstile-Mechanismus bezeichnet.
Der Bartell-Mechanismus ist eine Pseudorotation ähnlich dem Berry-Mechanismus und wurde erstmals an Iodheptafluorid IF7 mit pentagonal-pyramidaler Molekülgestalt beschrieben.[5] Hier erfolgt ebenfalls ein paarweiser Austausch von apikalen und äquatorialen Fluoratomen in der pentagonalen Bipyramide. Der Mechanismus weist dabei Charakteristiken der Berry-, Lever- und Turnstile-Pseudorotation auf.
Literatur und Quellen
- M. Binnewies, M. Jäckel, H. Willner, G. Rayner-Canham: Allgemeine und Anorganische Chemie. 1. Aufl., Spektrum, Heidelberg 2004, ISBN 978-3-82-740208-0, S. 488.
- M. E. Cass, K. K. Hii, H. S. Rzepa: Mechanisms that Interchange Axial and Equatorial Atoms in Fluxional processes: Illustration of the Berry Pseudorotation, the Turnstile and the Lever Mechanisms via animation of transition state normal vibrational modes. In: Journal of Chemical Education (Online). Bd. 83, Nr. 2, 2006, S. 336.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 782.
- R. R. Holmes: Structure of Cyclic Pentacoordinated Molecules of Main Group Elements. In: Phosphorus, Sulfur, and Silicon and the Related Elements. Bd. 98, Nr. 1–4, 1995, S. 205–221.
- J. E. Huheey, E. Keiter, R. L. Keiter: Anorganische Chemie. Prinzipien von Struktur und Reaktivität. 3. Aufl., de Gruyter, Berlin 2004, ISBN 978-3-11-017903-3, S. 275 ff.
- R. Luckenbach: Dynamic Stereochemistry of Pentacoordinated Phosphorus and Related Elements. 1. Aufl., Thieme, Stuttgart 1973, ISBN 3-13-456801-2.
- I. Ugi, D. Marquarding, H. Klusacek, P. Gillespie: Berry Pseudorotation and Turnstile Rotation. In: Accounts of Chemical Research. Bd. 4, Nr. 8, 1971, S. 288–296.
Einzelnachweise
- John E. Kilpatrick, Kenneth S. Pitzer, Ralph Spitzer: The Thermodynamics and Molecular Structure of Cyclopentane, Journal of the American Chemical Society 69, 2483–2488 (1947). doi:10.1021/ja01202a069.
- R. S. Berry: Correlation of Rates of Intramolecular Tunneling Processes, with Application to Some Group V Compounds. In: Journal of Chemical Physics. Bd. 32, Nr. 3, 1960, 32, S. 933–938.
- H. S. Gutowsky, D. W. McCall, C. P. Slichter: Nuclear Magnetic Resonance Multiplets in Liquids. In: Journal of Chemical Physics. Bd. 21, Nr. 2, 1953, S. 279–292.
- M. E. Cass, K. K. Hii, H. S. Rzepa: Mechanisms that Interchange Axial and Equatorial Atoms in Fluxional processes: Illustration of the Berry Pseudorotation, the Turnstile and the Lever Mechanisms via animation of transition state normal vibrational modes. In: Journal of Chemical Education (Online). Bd. 83, Nr. 2, 2006, S. 336.
- W. J. Adams, H. Bradford Thompson, L. S. Bartell: Structure, Pseudorotation, and Vibrational Mode Coupling in IF7: An Electron Diffraction Study. In: Journal of Chemical Physics. Bd. 53, Nr. 10, 1970, S. 4040–4046.