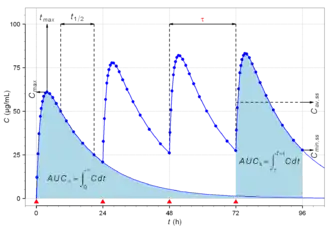
Pharmakokinetik
Die Pharmakokinetik beschreibt die Gesamtheit aller Prozesse, denen ein Arzneistoff im Körper unterliegt. Dazu gehören die Aufnahme des Arzneistoffes (Resorption), die Verteilung im Körper (Distribution), der biochemische Um- und Abbau (Metabolisierung) sowie die Ausscheidung (Exkretion). Ist zusätzlich vor der Resorption die Freisetzung (Liberation) des Arzneistoffes aus der Arzneiform von Bedeutung, wird auch die Abkürzung LADME für die Gesamtheit dieser Prozesse verwendet.
Als Begründer der Pharmakokinetik gilt der Kinderarzt Friedrich Hartmut Dost, der 1953 mit dem ersten Lehrbuch über die Pharmakokinetik Der Blutspiegel diese der klinischen Praxis und Forschung zugänglich machte. Grundlage seiner Überlegungen war die Erkenntnis, dass Dosisempfehlungen für Arzneimittel nicht einfach von Erwachsenen auf Kinder „heruntergerechnet“ werden dürfen. Aus diesen Grundüberlegungen entwickelte sich ein eigener Wissenschaftszweig, der heute ein wichtiger Bestandteil der Arzneimittelentwicklung ist.
Die Pharmakokinetik ist neben der Pharmakodynamik einer der beiden großen Teilbereiche der Pharmakologie. Bei der Pharmakokinetik geht es weitgehend um die Frage: Was macht der Organismus mit dem Wirkstoff? Bei der Pharmakodynamik geht es um die Frage: Was macht der Wirkstoff mit dem Organismus?
Übersicht
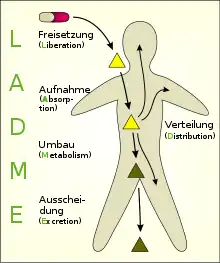
Neben der Freisetzung (Liberation) aus der Darreichungsform und der Aufnahme des Arzneistoffes in den Körper (Resorption, englisch: absorption) sind auch seine Verstoffwechselung im Organismus (Metabolisierung) und seine Ausscheidung maßgeblich für die Konzentration am Wirkort. Die aus dem Englischen abgeleitete Abkürzung LADME fasst diese Vorgänge zusammen:
- Liberation – Freisetzung
- Absorption – Aufnahme in die Blutbahn
- Distribution – Verteilung im Organismus
- Metabolism – Verstoffwechselung
- Excretion – Ausscheidung (renal, biliär, pulmonal, intestinal)
Freisetzung
Liegt der Arzneistoff nicht bereits in aufgelöster Form in der Arzneiform vor, so ist seine Freisetzung daraus der erste und oftmals geschwindigkeitsbestimmende Schritt im LADME-Prozess. Je nach therapeutischer Zielsetzung werden für den Wirkstoff unterschiedliche Freisetzungsprofile angestrebt.
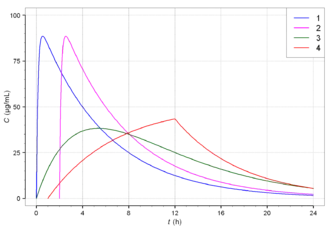
1 Schnell freisetzend
2 Verzögert freisetzend
3 Verlangsamt freisetzend, retardiert
4 Transdermales System
- Indikationen, die im Rahmen einer Behandlung mit festen Arzneiformen einen raschen Wirkungseintritt erlauben oder sogar erfordern (z. B. akute Schmerzen), werden mit schnell freisetzenden Tabletten oder Brausetabletten behandelt. Die schnelle Freisetzung kommt durch den raschen physikalischen Zerfall der Arzneiform zustande. Zäpfchen setzen den Arzneistoff etwas langsamer frei, da sie erst im Rektum schmelzen müssen, haben aber Vorteile in der Anwendung, wenn Übelkeit und Erbrechen die Therapie begleiten.
- Manche Indikationen erfordern eine modifizierte Abgabe des Arzneistoffes aus der Arzneiform, was nicht selten eine Herausforderung für seine technologische Formulierung (Galenik) darstellt. Durch die Wahl entsprechender Säure/Base-puffernder Hilfsstoffe, die pharmakologisch selbst keine therapeutische Wirkung besitzen, und durch die Herstellungstechnologie lässt sich die Kinetik der Freisetzung steuern und damit das Wirkungsprofil (Wirkstoffkonzentration und Wirkdauer) des Arzneimittels beeinflussen.
- Eine verzögerte Freisetzung aus einer peroralen Darreichungsform ist z. B. notwendig, wenn der Wirkstoff instabil gegenüber Magensäure ist. Ein säurefester Überzug sorgt dann dafür, dass die Tablette oder Kapsel den Wirkstoff erst nach der Magenpassage im neutralen oder leicht alkalischen Milieu des Dünndarms frei gibt.
- Eine verlangsamte Freisetzung (Retardierung) erlaubt die Verlängerung des Dosierungsintervalls und macht die Therapie anwendungsfreundlich (so ist beispielsweise statt 3 × 1 Tablette nur 1 × 1 Retardtablette täglich erforderlich). Zudem schwanken die Plasmaspiegel weniger und werden stattdessen auf einem gleich bleibenden Niveau gehalten.
- Therapeutische Systeme setzen den Arzneistoff besonders langsam und über einen langen Zeitraum kontrolliert frei. Sie kommen beispielsweise in Form von transdermalen Pflastern oder wirkstoffhaltigen Implantaten bzw. Inserten zum Einsatz.
Der Zusammenhang zwischen einer bestimmten Arzneiform und der Wirkung des enthaltenen Arzneistoffes ist Gegenstand der Biopharmazie.
Aufnahme
Unter Resorption versteht man die Aufnahme des Arzneistoffes vom Applikationsort in die Blutbahn. Je nach Arzneiform und Applikation geschieht dies vor allem über die Schleimhäute des Magen-Darm-Traktes (Tabletten, Säfte, Kapseln) einschließlich des Rektums (Zäpfchen) oder über die Haut (Salben, Cremes, Wirkstoffpflaster). Der Resorptionweg über die Alveolen wird in der Narkose mittels Inhalationsnarkotika genutzt. Bisher können nur wenige Arzneistoffe über die Nasenschleimhaut verabreicht werden (etwa Desmopressin, Oxytocin).
Dem Resorptionsvorgang liegen folgende Mechanismen zugrunde:
- passive Diffusion,
- Carrier-vermittelte Diffusion,
- aktiver Transport,
- Phagozytose bzw. Pinozytose.
Die Resorption wird durch zahlreiche Faktoren beeinflusst. Neben den chemisch-physikalischen Eigenschaften des Arzneistoffes sind besonders diese physiologischen Faktoren von Bedeutung:
- Größe und Zustand der Resorptionsfläche,
- Durchblutung an der Resorptionsfläche,
- Kontaktzeit mit der Resorptionsfläche.
Durchfallerkrankungen und der damit einhergehende beschleunigte Transport des Arzneistoffes durch den Magen-Darm-Trakt können aufgrund der knappen Kontaktzeit eine Resorptions- und Wirkminderung zur Folge haben (beispielsweise orale Kontrazeptiva, „Antibabypille“).
Verteilung
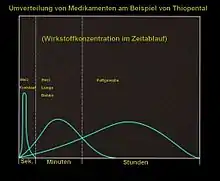
Sobald der Arzneistoff im Blutkreislauf zirkuliert, beginnt seine Verteilung (Distribution). In der Pharmakokinetik versteht man darunter den Stofftransport zwischen verschiedenen Körperflüssigkeiten und Geweben. Treibende Kraft des Transportvorgangs ist das Konzentrationsgefälle zwischen den verschiedenen Verteilungsräumen. Der Transportvorgang ist reversibel, d. h., er erfolgt „hin“ als auch wieder „zurück“.
Die Verteilung ist abhängig von:
- Substanzeigenschaften wie Löslichkeit, chemische Struktur und Bindungsvermögen an Plasmaproteine und Gewebsproteine. So neigen etwa lipophile Substanzen aufgrund ihrer guten Fettlöslichkeit besonders zu einer Anreicherung im Fettgewebe (Kumulation).
- Physiologischen Gegebenheiten wie die Organ- bzw. Gewebedurchblutung, dem pH-Wert im Gewebe bzw. in der Körperflüssigkeit und der Durchlässigkeit der zu durchdringenden Membranen. Eine schwer durchdringbare Membran umgibt beispielsweise die Hirnkapillaren und den Liquorraum (Blut-Hirn-Schranke, Blut-Liquor-Schranke) und verhindert oder vermindert auf diese Weise zentrale Nebenwirkungen.
Eine spezielle Form der Verteilung stellt der enterohepatische Kreislauf dar: der im Blut gelöste Arzneistoff bzw. seine Stoffwechselprodukte verteilen sich beim Passieren der Leber in die Galle, welche in den Darm abgesondert wird. Von dort werden die Substanzen wieder in die Blutbahn zurück resorbiert. Sie zirkulieren unter Umständen mehrfach und recht lange.
Metabolismus
Ein Arzneistoff unterliegt im Körper an verschiedenen Orten biochemischen Um- und Abbauprozessen, deren Gesamtheit als Metabolismus oder Biotransformation bezeichnet wird. Ziel dieser Prozesse ist dabei die Verbesserung der Ausscheidung aus dem Körper. Man unterscheidet dabei Phase-I-Reaktionen (Funktionalisierung) und Phase-II-Reaktionen (Hydrophilisierung). Die Reaktionsprodukte dieser Reaktionen bezeichnet man als Metaboliten eines Arzneistoffes.
Zu den Reaktionen der Phase I gehören beispielsweise Oxidationsreaktionen, Reduktionsreaktionen und die Hydrolyse. Durch diese Reaktionen nimmt die Wirkung eines Arzneistoffes im Allgemeinen ab. In bestimmten Fällen ist jedoch auch eine Wirkverstärkung bzw. der Umbau zu Metaboliten mit anderen Wirkungen möglich, wodurch es zu Nebenwirkungen kommen kann. Darüber hinaus gibt es, wie bereits erwähnt, mit den sogenannten Prodrugs auch Arzneistoffe, die erst durch die Metabolisierung aus einer wirkungslosen Form ihre beabsichtigte Wirkung erhalten. Zu den Phase-II-Reaktionen zählen vor allem die Glucuronsäure-Konjugation, die Aminosäure-Konjugation, die Sulfatierung und die Acetylierung. Im Ergebnis dieser Reaktionen wird ein Arzneistoff hydrophiler und damit wasserlöslicher, wodurch die Ausscheidung beschleunigt wird.
Hauptort der Metabolisierung ist die Leber. Darüber hinaus finden Metabolisierungsreaktionen auch in den verschiedenen Schleimhäuten, im Darm, in der Lunge und im Blutplasma statt. Während der Metabolisierung kann es zu Interaktionen zwischen verschiedenen gleichzeitig applizierten Medikamenten kommen. Dies ist beispielsweise dann möglich, wenn einer der beteiligten Arzneistoffe die Metabolisierungskapazität des Körpers auslastet, so dass die Metabolisierung eines zweiten Arzneistoffes verzögert wird. Dies führt für diesen Arzneistoff zu einem Anstieg der Wirkung. Solche Wechselwirkungen sind unter Umständen auch zwischen Medikamenten und bestimmten Inhaltsstoffen von Lebensmitteln möglich. Inhaltsstoffe von Grapefruit-Saft blockieren beispielsweise bestimmte Enzyme des Cytochrom-P450-Komplexes in der Leber. Dadurch wird der Abbau von vielen Medikamenten verzögert, so dass bei gleichzeitiger Einnahme von Grapefruit-Saft und entsprechenden Medikamenten deren Konzentration ansteigt, was zu Nebenwirkungen führen kann.
Auch die Wirkabnahme eines Medikaments bei längerer Einnahme ist auf die Metabolisierung zurückzuführen. In diesem Fall kommt es durch die wiederholte Einnahme zur sogenannten Enzyminduktion, einer verstärkten Bildung der an der Metabolisierung beteiligten Enzyme. Dadurch wird der betroffene Arzneistoff dann schneller abgebaut, wodurch seine Wirkdauer und -intensität, im Extremfall bis zur Wirklosigkeit, herabgesetzt werden. Erkrankungen an der Metabolisierung beteiligter Organe, insbesondere der Leber, können aufgrund der verringerten Metabolisierung zu einem Wirkanstieg und damit zu Nebenwirkungen führen. Für viele Metabolisierungsprozesse sind auch genetische Unterschiede bekannt. Diese können beispielsweise für bestimmte Reaktionen zu einer Unterscheidung zwischen Schnell- und Langsammetabolisierern führen.
Ausscheidung
Die Ausscheidung (Exkretion) eines Arzneistoffes bzw. seiner Metaboliten aus dem Blutkreislauf erfolgt zum größten Teil über die Nieren und den Urin (renale Ausscheidung). Ein geringer Teil wird über die Gallenflüssigkeit in den Dünndarm und im Weiteren mit dem Stuhl ausgeschieden. Wird die wirksame Substanz anschließend aus dem Darm wieder resorbiert (Rückresorption), so spricht man vom enterohepatischen Kreislauf.
Von untergeordneter Bedeutung ist die Exkretion über Haut (Schweiß) oder Schleimhaut (Darmschleimhaut, intestinale Ausscheidung), sowie über die Lunge (pulmonale Ausscheidung).
Mit der urinalen Ausscheidung von Ethinylestradiol, einem synthetischen Östrogen, welches in den meisten Antibabypillen enthalten ist, kommt es zu nachweisbaren Veränderungen bei Wasserlebewesen, wie zu einer Verlangsamung des natürlichen Fortpflanzungszyklus bei Fischen.[1] Die Exkretion von Arzneistoffen mit der Muttermilch kann zu Vergiftungserscheinungen beim gestillten Säugling führen.
Sonderfall Toxikokinetik
Die Toxikokinetik befasst sich mit der zeitlichen und quantitativen Konzentration eines Giftstoffes in verschiedenen Bereichen eines Organismus, z. B. in bestimmten Geweben.
Bei der Behandlung von Vergiftungen helfen Kenntnisse über die Toxikokinetik des Giftstoffes
- das Vergiftungsrisiko (die zu erwartende Schwere),
- die Notwendigkeit und Sinnhaftigkeit einer spezifischen Therapie, insbesondere der Verfahren zur Beschleunigung der Elimination
- sowie die Dauer und die Folgen der Vergiftung abzuschätzen.
Neben den Faktoren des LADME-Konzeptes sind hier zusätzlich noch folgende Faktoren zu beachten:
- Invasion (Resorption über Magen-Darm-Trakt, Haut und Schleimhäute, insbesondere der Atemwege)
- Speicherung (Bioakkumulation, als spezielle Form der Verteilung)
Kenngrößen
Wichtige beschreibende Parameter in der Pharmakokinetik sind zum Beispiel Verteilungsvolumen, Clearance, Bioverfügbarkeit, Plasmahalbwertszeit, Aufsättigungsdosis, Erhaltungsdosis.
Einflussgrößen
Einfluss auf das pharmakokinetische Verhalten eines Stoffes haben seine physikalisch-chemischen Eigenschaften und die biologischen Kenngrößen des Organismus.
- Physikalisch-chemische Parameter: Verteilungskoeffizient, Metabolische Umsatzrate, Löslichkeit, Dampfdruck, Diffusionsgeschwindigkeit, Proteinbindungskonstante
- Biologische Parameter: Alter, Geschlecht, Körpergewicht, Ausmaß körperlicher Aktivität, Anatomische Abmessungen, Blutfluss durch Organe, Organvolumina, Atmung
Relevanz
Pharmakokinetik in Arzneimittelzulassung
Genaue Kenntnisse zu allen genannten Prozessen sind essentieller Teil der für die Zulassung eines neuen Medikamentes notwendigen Antragsunterlagen. Entsprechende Daten werden in den jeweiligen Phasen der Entwicklung eines Medikamentes durch Studien gewonnen. Alle Prozesse des LADME beeinflussen den Konzentrations-Zeit-Verlauf und damit die Bioverfügbarkeit eines Medikamentes im Körper. Von Relevanz ist dies beispielsweise bei der Zulassung von Generika. Bei diesen ist vom Hersteller die sogenannte Bioäquivalenz zu demonstrieren, also die Vergleichbarkeit innerhalb bestimmter zulässiger Grenzen mit dem Originalpräparat hinsichtlich der Bioverfügbarkeit.
Therapeutische Aspekte der Pharmakokinetik
Die analytische Bestimmung der Arzneistoffkonzentration in Blut, Urin, Speichel und anderen Körperflüssigkeiten wird in der Therapie bei manchen Arzneimitteln angewendet, um die genaue Dosierung zu ermitteln und die Therapie zu verfolgen, insbesondere bei wiederholter und länger andauernder Therapie. Dies wird als Therapeutic Drug Monitoring bezeichnet.
Literatur
- Friedrich H. Dost: Der Blutspiegel. Kinetik der Konzentrationsabläufe in der Kreislaufflüssigkeit. Georg Thieme, Leipzig 1953, DNB 450986519 (362 S., 96 Abb.).
- Milo Gibaldi, Donald Perrier: Pharmacokinetics. In: Drugs and the Pharmaceutical Sciences. 2. Auflage. Volume 15. Marcel Dekker, Inc., New York, Basel 1982, ISBN 0-8247-1042-8 (englisch).
- Malcolm Rowland, Thomas N. Tozer: Clinical Pharmacokinetics. Concepts and Applications. 4. Auflage. Lippincott Williams & Wilkins, a Wolters Kluwer business, Baltimore, Philadelphia 2010, ISBN 978-0-7817-5009-7 (englisch).
- Hartmut Derendorf, Thomas Gramatte, Hans G. Schäfer: Pharmakokinetik. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart 2002, ISBN 3-8047-1907-4.
- Ernst Mutschler, Gerd Geisslinger, Heyo K. Kroemer, Peter Ruth, Monika Schäfer-Korting: Mutschler Arzneimittelwirkungen. Lehrbuch der Pharmakologie und Toxikologie. 9. Auflage. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart 2008, ISBN 978-3-8047-1952-1.
- Klaus Aktories, Ulrich Förstermann, Franz Hofmann, Wolfgang Forth: Allgemeine und spezielle Pharmakologie und Toxikologie. Urban & Fischer bei Elsevier, München und Jena 2004, ISBN 3-437-42521-8.
- Peter Langguth, Gert Fricker, Heidi Wunderli-Allenspach: Biopharmazie. Wiley-VCH, Weinheim 2004, ISBN 3-527-30455-X.
- E. J. Ariëns: Stereochemistry, a Basis for Sophisticated Nonsense in Pharmacokinetics and Clinical Pharmacology. In: European Journal of Clinical Pharmacology. Band 26, Nr. 6. Springer, 1984, S. 663–668, doi:10.1007/BF00541922.
- G. Gordon Gibson und Paul Skett: Introduction to Drug Metabolism, Chapman & Hall, London, Glasgow, New York, Tokyo, Melbourne, Madras 1992, ISBN 0-412-26390-4.
Weblinks
Einzelnachweise
- Karen Kidd: Effects of a Synthetic Estrogen on Aquatic Populations: a Whole Ecosystem Study. Freshwater Institute, Fisheries and Oceans Canada, Oktober 2004, archiviert vom Original am 19. Juni 2008; abgerufen am 5. September 2011 (englisch).