Konfokalmikroskop
Ein Konfokalmikroskop (von konfokal oder confocal, den gleichen Fokus habend) ist ein spezielles Lichtmikroskop. Im Gegensatz zur konventionellen Lichtmikroskopie wird nicht das gesamte Präparat beleuchtet, sondern zu jedem Zeitpunkt nur ein Bruchteil davon, in vielen Fällen nur ein kleiner Lichtfleck. Diese Beleuchtung wird Stück für Stück über das Präparat gerastert. Im Mikroskop entsteht also zu keinem Zeitpunkt ein vollständiges Bild. Die Lichtintensitäten des reflektierten oder durch Fluoreszenz abgegebenen Lichtes werden folglich nacheinander an allen Orten des abzubildenden Bereiches gemessen, so dass eine anschließende Konstruktion des Bildes möglich ist. Im Strahlengang des detektierten Lichts ist eine Lochblende angebracht, die Licht aus dem scharf abgebildeten Bereich durchlässt und Licht aus anderen Ebenen blockiert. Dadurch gelangt nur Licht aus einem kleinen Volumen um den Fokuspunkt zum Detektor, so dass optische Schnittbilder mit hohem Kontrast erzeugt werden, die fast nur Licht aus einer schmalen Schicht um die jeweilige Fokusebene enthalten.
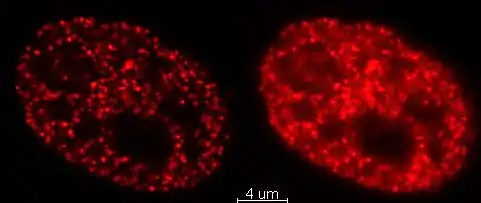
Heutige Konfokalmikroskope gibt es in verschiedenen Bauformen. Weit verbreitet sind Punktscanner, bei denen ein fokussierter Laserstrahl das Präparat abrastert (konfokales Laser-Scanning-Mikroskop, engl. confocal laser scanning microscope, CLSM, auch LSCM, nach engl. to scan: rastern). Bei Linienscannern wird dagegen eine ganze Bildzeile auf einmal erstellt, so dass eine höhere Geschwindigkeit erreicht werden kann. Eine dritte Variante benutzt eine Nipkow-Scheibe, auf der mehrere Lochblenden spiralförmig angeordnet sind. Bei der Rotation der Scheibe tastet jede Lochblende eine kreisförmige Kurve des Präparats ab. Diese Variante nutzt entweder Weißlicht zur Auflichtbeleuchtung, die zur Reflexion im Präparat führt. Oder es wird Fluoreszenz angeregt, wie es auch mit den anderen Bautypen möglich ist. Dann zählen sie zu den Fluoreszenzmikroskopen.
Erste Konfokalmikroskope wurden bereits Mitte des 20. Jahrhunderts gebaut. Es dauerte jedoch bis in die 1980er Jahre, bis neue technische Möglichkeiten, darunter Laser und Computersysteme, Weiterentwicklungen erlaubten, die zu größerer Verbreitung führten.
Das konfokale Prinzip
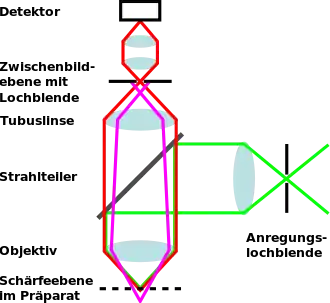
In einem Konfokalmikroskop wird eine punktförmige Lichtquelle in das Präparat abgebildet. Von der so beleuchteten Stelle wird Licht durch das Objektiv auf eine Lochblende fokussiert, bevor es den Detektor erreicht. Der Punkt in der Mitte der Lochblende und der Beleuchtungspunkt im Präparat sind dabei konfokal zueinander, das heißt, sie sind gleichzeitig im Fokus.
Konfokale Mikroskopie kann prinzipiell mit Durchlicht oder mit Auflicht realisiert werden. Heutige Konfokalmikroskope sind aber generell Auflichtmikroskope, sie benutzen das Objektiv für Beleuchtung und Detektion. Bei manchen Bautypen gibt es nicht nur einen, sondern mehrere Beleuchtungspunkte, die gleichzeitig an verschiedene Stellen des Präparats projiziert werden. Der Einfachheit halber wird das Prinzip hier am Beispiel eines einzigen Beleuchtungspunktes erläutert, der durch Auflicht erzeugt wird.
Strahlengang von der Lichtquelle zum Präparat
Das von der Lichtquelle kommende Licht (in der Schemazeichnung grün) wird zunächst in die Anregungslochblende fokussiert, um eine punktförmige Lichtquelle zu erzeugen. Auch im deutschen Sprachgebrauch werden diese und die zweite Lochblende häufig mit dem englischen Ausdruck Pinhole (wörtlich: Nadelloch) bezeichnet. In neueren konfokalen Laser-Scanning-Mikroskopen werden die Beleuchtungslaser mit Glasfasern eingekoppelt, und die Anregungslochblende kann dann durch diese ersetzt sein, da der kleine lichtführende Faserkern ähnliche optisch beschränkende Eigenschaften aufweist.
Die Beleuchtung wird auf einen Strahlteiler weitergeleitet. Bei Weißlichtmikroskopie wird ein halbdurchlässiger Spiegel eingesetzt, der einen ausreichenden Anteil der Beleuchtung zum Präparat spiegelt. Soll Fluoreszenz im Präparat nachgewiesen werden, wird ein dichroitischer Spiegel eingesetzt, der das Anregungslicht spiegelt, das Fluoreszenzlicht aber durchlässt. Schließlich wird durch das Objektiv im Präparat ein verkleinertes Bild der Anregungslochblende projiziert. Aufgrund der Beugung entsteht am Fokuspunkt ein Beugungsscheibchen (genauer: eine Punktspreizfunktion) und kein tatsächlicher Punkt.[1]
Strahlengang vom Präparat zur Detektionslochblende
Vom beleuchteten Punkt im Präparat geht das nachzuweisende Licht aus (rot in der Schemazeichnung). Dabei kann es sich um reflektiertes Licht oder um Fluoreszenz handeln. Der vom Objektiv aufgenommene Anteil durchtritt den Strahlteiler und wird in der Zwischenbildebene wieder in einem Punkt (einem Beugungsscheibchen) vereint. Bei modernen Objektiven mit unendlicher Tubuslänge ist hierfür noch eine Tubuslinse erforderlich. In der Zwischenbildebene ist die Detektions-Lochblende um diesen Punkt herum zentriert. Sie ist typischerweise gerade so groß, dass ihr Rand im ersten Minimum des Beugungsscheibchens verläuft. Der tatsächliche Durchmesser hängt also von der numerischen Apertur und der Vergrößerung des verwendeten Objektivs ab. Es kann auch noch eine weitere Vergrößerung bis zur Lochblendenebene vorgenommen werden, um den Durchmesser des Beugungsscheibchens und damit den Durchmesser der Lochblende zu vergrößern. Größere Blenden lassen sich einfacher fertigen.
In den meisten Präparaten wird Licht nicht nur vom tatsächlich beleuchteten Punkt im Präparat ausgesandt, sondern auch von Stellen darüber oder darunter (in der Schemazeichnung pink). Beispielsweise werden Fluoreszenzfarbstoffe auch in Ebenen über und unter der Schärfeebene angeregt. Das von diesen Punkten kommende Licht wird jedoch nicht in der Zwischenbildebene, sondern in davor oder dahinter liegenden Ebenen zu einem Punkt vereint, so dass die Lichtstrahlen von diesen Punkten in der Zwischenbildebene als Strahlkegel vorliegen. Der überwiegende Teil dieser Strahlkegel wird deshalb durch die Lochblende blockiert, so dass am Detektor nur sehr wenig Licht ankommt, das nicht vom Fokuspunkt im Präparat ausgesandt wurde.
Optische Information, die nicht aus dem Fokuspunkt des Präparats kommt, wird somit doppelt unterdrückt: Erstens wird sie nicht „abgefragt“, da die Beleuchtungsintensität außerhalb des Fokus schwach ist, und zweitens wird Licht von außerhalb des Fokuspunkts an der Lochblende fast vollständig blockiert. Dadurch werden eine deutliche Kontrastverbesserung und auch eine etwas bessere Auflösung erzielt.
Der sogenannte „Fokuspunkt“, aus dem das Licht zur Bildentstehung beiträgt, ist ein dreidimensionales Volumen. Die genaue Form dieses Volumens wird als Punktspreizfunktion (engl. point spread function, abgekürzt PSF) bezeichnet. Je kleiner das Volumen ist, desto besser ist die Auflösung des Mikroskops.
Da nur aus einem kleinen Volumen des Präparats Licht zum Detektor gelangt, ist es für die Bilderzeugung notwendig, dieses Volumen über das Präparat zu bewegen, also die Probe abzurastern. Auch wenn mehrere Fokuspunkte eingesetzt werden, müssen diese über das Präparat bewegt werden. Im Mikroskop selbst entsteht also zu keinem Zeitpunkt ein komplettes Bild des Präparats, dieses wird erst anschließend im Computer erzeugt oder, im Fall von mehreren Punkten, auf dem Film oder dem Chip einer Kamera.
Das konfokale Laser-Scanning-Mikroskop – Abrastern mit einem fokussierten Laserstrahl
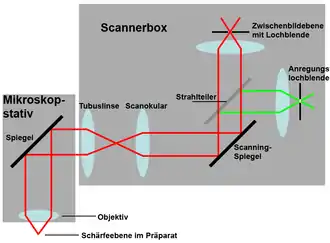
In der biomedizinischen Forschung sind konfokale Laser-Scanning-Mikroskope (Abk. CLSM, nach englisch: confocal laser scanning microscope, seltener LSCM, deutsch auch: Laserrastermikroskop) weit verbreitet, bei denen ein vom Objektiv fokussierter Laserstrahl ein Objekt punktweise abrastert. Meistens wird dabei Fluoreszenz von speziellen Markern nachgewiesen, es handelt sich dann um eine Form der Auflicht-Fluoreszenzmikroskopie. Laser werden eingesetzt, da von anderen Lichtquellen nicht genügend Licht für eine intensive Fluoreszenzanregung auf einen Punkt konzentriert werden kann. Mit derartigen Geräten kann auch konfokale Reflexionsmikroskopie betrieben werden.[1]
Die für die konfokale Mikroskopie zusätzlich erforderlichen optischen Elemente werden bei kommerziellen Geräten in einer Box untergebracht, die an ein qualitativ hochwertiges Mikroskop angeflanscht wird. Das Laserlicht wird in diese Scannerbox geleitet und von dort in das Mikroskop gespiegelt, um durch das Objektiv ins Präparat zu gelangen. Das im Präparat erzeugte Signal geht den umgekehrten Weg zurück zur Box, wo hinter der Lochblende die Signalintensität gemessen wird. Der Aufnahmevorgang wird von einem Computer gesteuert und die Bilder werden am Computermonitor angezeigt.
Vom Laser zum Präparat
.jpg.webp)
Typischerweise haben diese Geräte mehrere Anregungslaser, zum Beispiel einen Argonlaser, der mehrere Wellenlängen emittiert (458, 488, 514 nm und andere), und Helium-Neon-Laser, die Licht mit 543 oder 633 nm aussenden. Das Licht verschiedenfarbiger Laser kann mit dichroitischen Strahlteilern übereinandergelegt werden. In vielen aktuellen Geräten wird die Intensität der jeweiligen Wellenlängen über einen akusto-optischen Modulator (auch: Acousto-Optical Tunable Filter, AOTF) moduliert.
Bei neueren Geräten wird das Laserlicht häufig über Glasfasern zur Scannerbox geleitet. Da der kleine lichtführende Kern einer Glasfaser optisch ähnlich wirkt wie eine Lochblende kann die Anregungslochblende entfallen. Das Licht fällt nun auf den dichroitischen Strahlteiler, der es zum ersten Scanspiegel lenkt. Bei der Verwendung von mehreren Anregungswellenlängen können Doppel- oder Tripel-Strahlteiler eingesetzt werden, die zwei oder drei Wellenlängen zum Präparat hin spiegeln, das jeweilige Fluoreszenzlicht aber durchlassen. Bei manchen Geräten übernimmt diese Funktion ein weiterer akusto-optischer Modulator, der dann als Acousto-Optical Beam Splitter (AOBS) bezeichnet wird.
Die Bewegung der Scanspiegel bestimmt, wie schnell und in welchem Bereich der Anregungspunkt (genauer: das beugungsbegrenzte Anregungsvolumen) über das Präparat rastert. Die Scangeschwindigkeit wird in Bildzeilen pro Sekunde, also in Hertz (Hz) angegeben. Typische Geschwindigkeiten liegen zwischen 200 und 2000 Hz. Durch eine genau gesteuerte Auslenkung der Spiegel werden die Größe und die Position des abgerasterten Bereiches festgelegt. Der Anregungspunkt wird innerhalb einer Bildzeile kontinuierlich über das Präparat bewegt und durch Festlegung der sogenannten pixel dwell time (Pixelverweildauer, Zeit bis das Signal dem nächsten Pixel zugeordnet wird) ergibt sich die Anzahl der Bildpunkte (Pixel) pro Zeile. Zusammen mit der Anzahl der Bildzeilen ergibt sich die Gesamtzahl der Bildpunkte, zum Beispiel 512 × 512 Pixel. Eine variable Ansteuerung der Scanspiegel erlaubt es daher, mit dem gleichen Objektiv unterschiedlich stark vergrößerte Bilder aufzunehmen. Dies ist ein wichtiger Unterschied zu Kamera-basierten Systemen.
Durch weitere Linsen und Spiegel wird das Anregungslicht schließlich durch das Objektiv auf das Präparat geleitet, wo es zur Fluoreszenzanregung oder zur Reflexion kommt.
Vom Präparat zum Detektor

Das Fluoreszenzlicht nimmt den gleichen Weg zurück über die Scanspiegel, passiert den dichroitischen Strahlteiler und gelangt gemäß dem oben beschriebenen konfokalen Prinzip zur Lochblende in einer Zwischenbildebene und schließlich zu den Detektoren. Alternativ oder zusätzlich kann auch reflektiertes Licht über diesen Strahlengang aufgefangen werden. Neben dem Auflösungsvermögen des verwendeten Objektivs (genauer: seiner numerischen Apertur) und der Wellenlänge des jeweiligen Lichts bestimmt der Durchmesser der Lochblende die Tiefenschärfe und damit die „Dicke“ des optischen Schnittes. Liegt der Durchmesser der Lochblende im ersten Minimum des Beugungsscheibchens (1 Airy unit), so wird das meiste Licht aus anderen Ebenen blockiert und der größte Teil des eigentlichen Signals tritt durch. Wird die Blende stärker geöffnet, so tritt mehr Licht aus höher und tiefer gelegenen Präparateebenen durch, so dass mehr unscharfe Anteile zum Bild beitragen (siehe Abbildungen). Wird die Blende stärker geschlossen als eine Airy unit, so tritt ein starker Helligkeitsabfall ein, der ein deutliches Bild ebenfalls erschwert.[1][2]
Um mehrere Fluoreszenzfarben oder reflektiertes und Fluoreszenzlicht parallel konfokal aufnehmen zu können, wird das Licht vor dem Detektor spektral aufgetrennt. Theoretisch müsste die Detektionslochblende für jede Wellenlänge in der Größe angepasst werden, da der Durchmesser des Beugungsscheibchens linear von der Wellenlänge abhängt. Tatsächlich geschah die spektrale Auftrennung in manchen früheren Geräten (zum Beispiel im Zeiss LSM 410) zuerst und jeder Farbbereich hatte anschließend seine eigene Lochblende. Aus praktischen Gründen verwenden heutige kommerzielle Geräte jedoch nur eine Lochblende für alle Farben. Die spektrale Auftrennung geschieht erst dahinter, beispielsweise mit dichroitischen Strahlteilern, die verschiedene Farbanteile auf unterschiedliche Detektoren lenken.
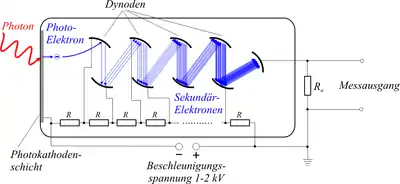
Als Detektoren werden in kommerziellen Geräten meistens Photomultiplier (PMTs) und bei speziellen Anwendungen manchmal auch Avalanche-Photodioden (APDs) eingesetzt.[1] Neue Hybrid-Photodetektoren (HPDs) verbinden Eigenschaften von PMTs und APDs. Sie werden, wie auch viele normale PMTs, von Hamamatsu Photonics hergestellt[3] und von verschiedenen Mikroskopanbietern eingebaut.[4][5][6]
Manche Geräte haben einen weiteren Detektor im Strahlengang hinter dem Präparat, der durchtretendes Laserlicht auffängt. Im Computer kann aus den Messwerten eine Art Hellfeldbild rekonstruiert werden, in dem Licht-absorbierende oder ablenkende Strukturen im Präparat durch dunkle Stellen repräsentiert werden. Das Licht tritt auf dem Weg zu diesem Detektor aber nicht durch eine Lochblende, so dass kein konfokales Bild entsteht.
Besonderheiten der Aufnahme
Um das Signal-Rausch-Verhältnis und damit die Bildqualität zu erhöhen, erlaubt es die Steuersoftware, ein Bild mehrmals aufzunehmen und den Mittelwert zu bilden. Besonders bei schwach fluoreszenten Präparaten hilft dies, den Einfluss des Poisson-Rauschens der aufgefangenen Photonen und des statistischen Rauschen der elektronischen Komponenten auf das Bild zu reduzieren, da sich Rauschen bei jeder Aufnahme anders verteilt und somit bei Mehrfachaufnahmen weniger ins Gewicht fällt.
Heutige kommerzielle konfokale Laser-Scanning-Mikroskope können durch Bewegung des Objektivs oder des Präparats die Schärfeebene stufenweise verschieben, um optische Serienschnitte zu erzeugen (siehe Filmsequenz). Derartige Bildserien können als Grundlage für dreidimensionale Computerrekonstruktionen verwendet werden (siehe Abbildungen).
Punktscanning-Verfahren ohne Scanspiegel
Frühe konfokale Mikroskope hatten einen unbeweglichen Strahlengang, stattdessen wurde das Präparat bewegt, da dies technisch einfacher zu realisieren war (siehe unten, Geschichte). Zwar ist dieser „Stage-Scanning“-Ansatz (von engl. stage für Objekttisch) auf Grund der zu bewegenden Masse deutlich langsamer, er hat jedoch den Vorteil, dass die Beleuchtungsintensität für jede Stelle des Präparats exakt gleich ist. Bei „Beam-Scanning“, also der oben dargestellten Bewegung des Laserstrahls über das nicht bewegte Präparat, ist dagegen die Beleuchtung zum Rand des Gesichtsfeldes etwas weniger intensiv. Bei Anwendungen, für die dies wichtig und eine hohe Geschwindigkeit nicht erforderlich ist, kann daher auch heute (Stand 2013) Stage-Scanning zum Einsatz kommen, so bei der Fluoreszenzkorrelationsspektroskopie (FCS).[7] Bei FCS wird die Fluoreszenzintensität im detektierten Volumen über längere Zeit an einem Punkt gemessen, so dass zwar Konfokalität, aber keine Rastervorrichtung zwingend erforderlich ist.
Eine dritte Möglichkeit, um eine Ebene des Präparats abzurastern, besteht in der seitlichen Verschiebung des Objektivs. Diese Möglichkeit wird selten angewendet. Das erste konfokale Mikroskop mit Laser verwendete diesen Ansatz (siehe unten),[8] aber auch aktuelle (2017) kommerzielle Geräte sind mit dieser Option erhältlich.[9]
Kommerzielle Geräte
Konfokale Laser-Scanning-Mikroskope mit Beam-Scanning für die Lebenswissenschaften werden von allen vier großen Mikroskopherstellern angeboten (Carl Zeiss, Leica Microsystems, Nikon und Olympus). Manche dieser Geräte verbinden konfokale Mikroskopie mit anderen Laser-Scanning-Mikroskopischen Anwendungen wie Multiphotonenmikroskopie oder STED-Mikroskopie. Bio-Rad, der erste Hersteller von kommerziellen konfokalen Laser-Scanning-Mikroskopen, hat seine Mikroskopabteilung zwischenzeitlich an Zeiss verkauft, so dass die Marke im Mikroskopmarkt nicht mehr auftritt. Daneben gibt es eine Reihe von Herstellern, die konfokale Laser-Scanning-Mikroskope für spezielle Anwendungen anbieten. Dazu gehören PicoQuant,[9] HORIBA,[10] ISS[11] und WITec.[12]
Spezielle Anwendungen mit konfokalen Laser-Scanning-Mikroskopen
Die besonderen Eigenschaften des konfokalen Laser-Scanning-Mikroskops erlauben neben der Herstellung von optischen Schnitten auch weitere Anwendungen. Teilweise ist dafür eine zusätzliche Ausstattung erforderlich. Zu diesen Techniken gehören Fluorescence Recovery after Photobleaching (FRAP), Fluoreszenzkorrelationsspektroskopie (FCS), Einzelmolekülfluoreszenzspektroskopie, Förster-Resonanzenergietransfer (FRET), Raman-Spektroskopie und Fluoreszenzlebensdauer-Mikroskopie (FLIM).
Linienscanner
Punktscanner sind relativ langsam, da jeder Punkt im Präparat einzeln abgerastert werden muss. Dies ist nicht nur ein technisches Problem der erreichbaren maximalen Geschwindigkeit: Bei einem zu schnellen Scanvorgang der einzelnen Bildpunkte wird auch nicht genügend Fluoreszenzlicht aufgefangen, um ein Bild mit ausreichendem Kontrast zu erstellen. Eine Möglichkeit, dieses Problem zu umgehen, ist, das Präparat mit einer Linie (statt mit einem Punkt) zu beleuchten und das Fluoreszenzlicht statt durch eine Lochblende durch eine entsprechende Schlitzblende zu führen. Daraus ergibt sich eine unterschiedliche (anisotrope) Auflösung in x- und y-Richtung. In Richtung der Scanlinie und der Schlitzblende (x) entspricht die Auflösung einem konventionellen Mikroskop, die Vorteile des Konfokalmikroskops kommen nur noch senkrecht dazu (y-Richtung) zum Tragen. Vor der Entwicklung von Spinning-Disk-Mikroskopen, die für die Fluoreszenzmikroskopie geeignet sind, waren Linienscanner die mit Abstand schnellste Möglichkeit, konfokale Bilder von schwach fluoreszierenden Präparaten zu erstellen.[13]
Die technische Hauptschwierigkeit beim Bau und Betrieb von Linienscannern ist die Realisierung einer beugungsbegrenzten schmalen Anregungslinie mit gleichmäßiger Helligkeit und einer ausreichend schmalen Schlitzblende sowie die Ausrichtung der beiden exakt parallel zueinander. Die Ausrichtung erfordert dabei nicht nur wie beim Punktscanner Bewegung in x- und y-Richtung, sondern auch Rotation. Wenn die beiden Kanten der Schlitzblende nicht völlig gleichmäßig und parallel zueinander sind, kann dies zu Streifen im Bild führen, beispielsweise wenn sich Staub an der Blendenkante anlagert. Diese Probleme führten in der Praxis dazu, dass sowohl Anregungslinie als auch Schlitzblende erheblich breiter waren, als sie theoretisch sein sollten.[13]
Die Linie des Fluoreszenzlichtes kann entweder mit einem CCD-Zeilen-Detektor aufgefangen werden (bei den Geräten LSM5 live und Duo von Zeiss), oder die Linie wird über einen beweglichen Spiegel auf eine Kamera abgebildet, so dass auf dem Kamerachip ein Bild zeilenweise aufgebaut wird (Meridian Insight, BioRad DVC 250). Aufgrund der hohen Scangeschwindigkeit lässt sich dieses Bild auch über ein Okular mit dem Auge betrachten.[13]
Statt einer Linie kann das Präparat auch mit mehreren nebeneinander liegenden Linien beleuchtet werden. Das für die Oberflächenuntersuchung von Werkstoffen vorgesehene Zeiss CSM 700 verwendet eine Schlitzmaske im Beleuchtungsstrahlengang, um auf dem Präparat ein Streifenmuster zu erzeugen. Zur Detektion wird die Fokusebene auf dem Chip einer Kamera abgebildet, die Funktion der Schlitzblende wird digital nachgebildet, indem nur bestimmte Pixel ausgelesen werden. Durch verschieben der Schlitzmaske wird das Präparat schließlich vollständig erfasst. Da weißes Licht zur Anregung verwendet wird, können Bilder in Echtfarben aufgenommen werden. Die berührungsfreie Untersuchung von Oberflächen wird ermöglicht, indem nur mit Trockenobjektiven gearbeitet wird.[14]
Konfokale Mikroskope mit Nipkow-Scheibe – Abrastern mit vielen fokussierten Lichtstrahlen

Eine weitere Möglichkeit zur schnellen konfokalen Aufnahme ist die Verwendung von vielen, parallel genutzten Lochblenden auf einer Nipkow-Scheibe. Der Name ist etwas irreführend: Die Scheibe, die Paul Nipkow im 19. Jahrhundert zur Übertragung von Fernsehbildern entwickelte, enthielt eine Spirale mit Löchern. Zur konfokalen Mikroskopie verwendete Nipkow-Scheiben enthalten dagegen viele, dicht nebeneinander liegende Spiralarme. Die Beleuchtung trifft auf die Scheibe und tritt teilweise durch die Lochblenden hindurch. Diese werden in das Präparat verkleinert, um viele Fokuspunkte zu erzeugen.
Durch Drehung der Scheibe rastern die Fokuspunkte in Kreisbögen sehr schnell über das Präparat, so dass im Bruchteil einer Sekunde ein vollständiges Bild entsteht. Dadurch kann das Bild auch mit dem Auge erkannt werden. Als Detektor wird eine Kamera eingesetzt, in der minimalen Belichtungszeit wird der untersuchte Präparateausschnitt einmal abgerastert.[13]
Für Fluoreszenzmikroskopie waren Mikroskope mit Nipkow-Scheiben zunächst wenig geeignet, da die Löcher der Nipkow-Scheibe weniger als ein Prozent der Beleuchtung durchlassen. Dadurch ist die Anregung für die allermeisten fluoreszenzmarkierten Präparate zu schwach. Zwar gibt es entsprechende Geräte, die für den Bereich der Lebenswissenschaften angeboten werden, sie sind jedoch wenig verbreitet. Erst etwa ab der Jahrtausendwende wurde die Anregungsstärke durch neue technische Ansätze verbessert.[13]
Tandem-Scanner für die Weißlichtreflexionsmikroskopie
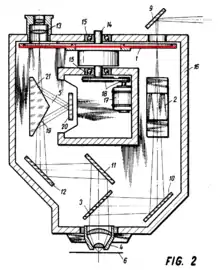
In den 1960er Jahren entwickelte der Tschechoslowake Mojmír Petráň das „Tandem-Scanning-Mikroskop“ (TSM), das konfokale Reflexionsmikroskopie ermöglichte. Es heißt so, weil der Beleuchtungsstrahlengang durch eine Seite der Nipkow-Scheibe geht und der bildgebende Strahlengang durch die gegenüberliegende Seite. Die Lochblenden auf beiden Seiten rastern also bei der Drehung der Scheibe im Tandem. Jeder Beleuchtungslochblende entspricht eine Detektionslochblende auf der genau gegenüberliegenden Position.[13][15]
Für die ursprüngliche Version des Mikroskops wurde die Nipkow-Scheibe von 8,5 cm Durchmesser aus 20 Mikrometer dünner Kupferfolie hergestellt, in die 26.400 Löcher von etwa 90 Mikrometer Durchmesser und durchschnittlich 280 µm Abstand von Lochmitte zu Lochmitte geätzt wurden. Die Löcher waren in 80 archimedischen Spiralen angeordnet. Die Scheibe rotierte dreimal pro Sekunde, das Beobachtungsfeld wurde 120-mal pro Sekunde abgerastert. Beleuchtungsquelle war eine Wolframlampe oder, zur stärkeren Beleuchtung, ein Bild der Sonne.[16][17][18]
Das Licht, das durch die Nipkow-Scheibe (in der Zeichnung rot unterlegt) tritt gelingt nach mehreren Spiegelungen zu einem teildurchlässigen Spiegel (über dem Objektiv, 3), der einen Teil der Beleuchtung zum Objektiv reflektiert. Der durch diesen Spiegel durchtretende Anteil der Beleuchtung geht verloren. Das Objektiv bildet die Lochblenden im Präparat ab. Das an diesen Stellen reflektierte Licht wird vom Objektiv wieder aufgenommen und zum teildurchlässigen Spiegel geleitet. Diesmal ist nur der durchtretende Anteil von Interesse: Er wird durch mehrere Spiegelungen zur gegenüberliegenden Seite der Nipkow-Scheibe geleitet. Durch eine anspruchsvolle Fertigungstechnik wird sichergestellt, dass beide Hälften der Scheibe optisch identisch sind,[18] so dass nun entsprechend dem konfokalen Prinzip das in den Beleuchtungspunkten reflektierte Licht aus der Fokusebene durch die Lochblenden durchtreten kann.
Theoretisch ergibt die Beleuchtung mit weißem Licht ein Echtfarbenbild. Auch sehr gute Glasoptiken können aber chromatische Aberration nicht ganz vermeiden. Dadurch ist die Beleuchtung im Präparat für verschiedene Farben in etwas verschobenen Ebenen.[13]
In der ersten Veröffentlichung zu Petráňs Mikroskop wurde die Abbildung von Spinalganglien und Gehirnen beschrieben.[16] Tandem-Scanning-Mikroskope werden auch heute noch eingesetzt, beispielsweise bei der Untersuchung der Hornhaut des Auges.[15]
Einseitige Nipkow-Scheiben-Mikroskope für Weißlichtreflexionsmikroskopie
 Reflexionsdaten einer 3D-Oberflächenvermessung einer Ein-Euro-Münze mit einem konfokalen Weißlichtmikroskop; dargestellt ist einer der erhabenen Sterne. |
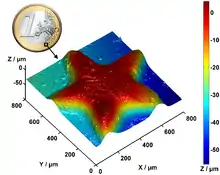 3D-Profil, gewonnen aus den Bilddaten links |
Im Gegensatz zum Tandemscanner wird die Nipkow-Scheibe hier nur auf einer Seite durchstrahlt: Dieselben Lochblenden kommen erst bei der Beleuchtung und dann bei der Detektion zum Einsatz.
Von oben kommend trifft die Beleuchtung zunächst auf einen Strahlteiler, der einen Teil des Lichts zur Nipkow-Scheibe leitet. Der hier durchtretende Anteil gelangt durch das Objektiv zum Präparat, wo korrespondierend zu jeder Lochblende je ein Fokuspunkt entsteht. Reflektiertes Licht fällt zurück ins Objektiv und gelangt durch dieselbe Lochblende weiter Richtung Strahlteiler. Dort wird es teilweise zur Kamera gespiegelt, der Rest geht verloren.[13]
Dabei gilt es zu vermeiden, dass von der Nipkow-Scheibe selbst reflektiertes Licht zur Kamera gelangt. Dies kann erreicht werden, indem die Nipkow-Scheibe verspiegelt und ein wenig geneigt wird. Allerdings befinden sich die Lochblenden dann nicht mehr in der optimalen Position. Um dies auszugleichen, müssen sie etwas größer gemacht werden. Alternativ kann mit polarisiertem Licht gearbeitet werden.[13]
Derartige Mikroskope für Reflexionsbilder werden in den Materialwissenschaften eingesetzt, um Oberflächen zu untersuchen, beispielsweise in der Halbleiterindustrie. Hier sind sie besser geeignet als konfokale Laser-Scanning-Mikroskope, da das kohärente Licht eines Lasers bei Reflexion auf glatten Oberflächen zu unerwünschten Interferenz-Effekten führt, die bei Beleuchtung mit nicht kohärentem Weißlicht vermieden werden.[13][19]
Eine zweite Scheibe mit Mikrolinsen
Die japanische Firma Yokogawa entwickelte als erste ein konfokales Nipkow-Scheiben-System, das für Fluoreszenzanregung geeignet ist, die Yokogawa-Spinning-Disk. Über der Nipkow-Scheibe befindet sich eine zweite Scheibe, die synchron mitdreht und auf der Mikrolinsen aufgebracht sind. Jeder Lochblende ist dadurch eine Mikrolinse vorgeschaltet, die das Licht auf die Öffnung fokussiert. So wird über 60 % zum Präparat durchgelassen. Der für die Fluoreszenzmikroskopie erforderliche dichroitische Strahlteiler befindet sich zwischen den beiden Scheiben und koppelt die Fluoreszenz, die vom Präparat zurückkommt, seitlich aus, Richtung Kamera. Dadurch geht das Fluoreszenzlicht zwar durch die Lochblenden, aber nicht durch die Mikrolinsenscheibe, so dass Lichtverluste hier vermieden werden. Außerdem kann Streulicht, das im Anregungsstrahlengang beim Auftreffen auf die Mikrolinsenscheibe entsteht, nicht zur Kamera gelangen.[13]
Die gute Lichtausbeute des Systems mit etwa 2000 gleichzeitig genutzten konfokalen Lochblenden ermöglicht Echtzeitbeobachtungen mit deutlich besserem Signal-zu-Rausch-Abstand als typische konfokale Punktscanner bei einer vergleichbaren Anzahl von Bildern pro Sekunde. Da ein Bildpunkt nicht nur einmal, sondern häufiger hintereinander abgerastert werden kann, ist die maximale Lichtpunktbelastung im Präparat niedriger als in typischen Punktscannern, so dass die Fluorochrome im Präparat weniger stark ausbleichen. Die Scheiben drehen sich mit 5 oder 10 Umdrehungen pro Sekunde. Einige publizierte Arbeiten haben Bildraten von 15 Bildern pro Sekunde erreicht. Durch die redundanten Muster auf den Scheiben ist weniger als eine Scheibenumdrehung erforderlich, um ein vollständiges Bild aufzunehmen. Auf Grund der hohen Bildraten ist es im Gegensatz zum Punktscanner auch möglich, das konfokale Bild durch das Okular mit dem Auge zu betrachten.[13]
Yokogawa selbst bietet keine Mikroskope an. Stattdessen wird das beschriebene Bauteil von anderen Firmen in eigene Geräte eingebaut, beispielsweise von Leica Microsystems, PerkinElmer und Zeiss. Als Lichtquelle werden Laser eingesetzt, da diese eine gleichmäßige und starke Beleuchtung ermöglichen. Daher handelt es sich bei diesem Gerätetyp auch um konfokale Laser-Scanning-Mikroskope. Es hat sich aber die Bezeichnung „Spinning Disk“-Mikroskope durchgesetzt.[13]
Eine Scheibe mit Hohlspiegeln
Einen alternativen Ansatz für eine gute Lichtausbeute zur Fluoreszenzanregung mit Nipkow-Scheiben entwickelte Till Photonics in Gräfelfing. Bei diesem „Andromeda“-System wird Laserlicht über einen sogenannten Corner Cube eingespeist. Durch diesen geht das Licht zunächst gerade durch, wird vom dichroitischen Strahlteiler gespiegelt und geht weiter durch eine Linse zur Nipkow-Scheibe. Auf der Scheibe ist jede der vielen Lochblenden von einem sechseckigen konkaven Spiegel umgeben. Nur wenig Licht tritt sofort durch die Lochblenden durch. Der Rest wird zurückgespiegelt und trifft nach dem dichroitischen Strahlteiler wieder auf den Corner Cube. Von dieser Seite wirkt der Corner Cube wie ein flacher Spiegel, so dass das Licht wieder zurück zur Nipkow-Scheibe geleitet wird. Bei diesem zweiten Durchlauf tritt nun auf Grund der Wirkung der konkaven Spiegel der Großteil des Lichtes durch die Lochblenden durch und trifft auf das Präparat. Das vom Präparat zurückkommende Fluoreszenzlicht tritt dem konfokalen Prinzip entsprechend durch die Nipkow-Scheibe hindurch weiter zum dichroitischen Strahlteiler und durch diesen schließlich zur Kamera. Ein Vorteil dieses Systems ist, dass die dichroitischen Strahlteiler in einem Unendlich-Bereich des Strahlengangs liegen. Dadurch sind deren Charakteristika weniger kritisch für die Bildqualität.[20]
Vor- und Nachteile von konfokalen Nipkow-Scheiben-Systemen im Vergleich zu Punktscannern
Durch die gleichzeitige Verwendung von vielen Lochblenden und damit vielen Beleuchtungspunkten können auf Nipkow-Scheiben basierende Systeme ein Präparat schneller abrastern als ein Punktscanner. Allerdings geht in einem konventionellen Nipkow-Scheiben-System der Großteil der Beleuchtung an der Scheibe verloren. Während die durchtretende Lichtmenge bei konfokalen Weißlichtmikroskopen trotzdem ausreicht, sind für die Fluoreszenzmikroskopie lichterhaltende Vorrichtungen erforderlich. Dann aber erlauben derartige Systeme ein schonenderes Aufnehmen von lebenden fluoreszenten Zellen, da die maximale Lichtbelastung jedes Punktes niedriger ist (siehe oben).
Durch die Verwendung mehrerer nebeneinander liegender Lochblenden in der Nipkow-Scheibe ist es möglich, dass Licht von einem Punkt im Präparat durch die falsche Lochblende zur Kamera gelangt und so die Konfokalität des Bildes einschränkt. Bei Materialuntersuchungen von Oberflächen tritt dieser Effekt kaum auf. Bei dickeren biologischen Präparaten, in denen es zu Streuung kommt, kann dies jedoch zum Problem werden. Der konfokale Effekt wird dann mit zunehmender Tiefe der Schärfeebene abgeschwächt.[13]
Ferner ist bei Nipkow-Scheiben-Systemen die beobachtbare Region im Präparat fest vorgegeben. Ein Herein- oder Herauszoomen, also eine Änderung der Größe der abgerasterten Region wie beim Punktscanner, ist nicht möglich.[13] Auch dies ist bei Materialuntersuchungen unproblematisch, da die Proben nicht ausbleichen und somit eine verkleinerte abgerasterte Region keinen Vorteil darstellt. Auch kann hier eine höhere Vergrößerung durch Wechsel zwischen den Trockenobjektiven oder durch eine vergrößernde Optik direkt vor der Kamera erzielt werden. Dagegen sind für Lebendzellbeobachtungen verwendete Spinning-Disk-Fluoreszenzmikroskope in der Regel von inverser Bauart und es werden Immersionsobjektive eingesetzt, da diese auf Grund der höheren numerischen Apertur einen größeren Anteil der Fluoreszenz auffangen. Da die Immersionsflüssigkeit bei Objektivwechsel entfernt und neu aufgetragen werden muss, ist ein Objektivwechsel hier aufwändiger. Zusätzliche Zoomoptiken führen immer auch zu etwas Lichtverlust durch Spiegelungen an den zusätzlichen Glasoberflächen, daher wird auf diese bei lichtschwachen Fluoreszenzbeobachtungen zugunsten einer höheren Sensitivität verzichtet.
Unterschiedliche Objektive produzieren unterschiedlich große Beugungsscheibchen, je nach Vergrößerung und numerischer Apertur. Die Lochblenden auf einer Scheibe sind aber in der Größe nicht veränderbar, so dass die optimale Größe nur für ein einziges Objektiv oder für wenige Objektive erreicht werden kann. Bei Punktscannern kann die Größe der Lochblende dagegen an das jeweils verwendete Objektiv und an die verwendete Anregungswellenlänge angepasst werden.
Einseitige Nipkow-Scheiben-Systeme haben dafür den Vorteil, dass eine Justierung von Anregungs- und Emissionslochblende zueinander wie bei Punktscannern nicht erforderlich ist, da für beide Zwecke derselbe Satz von Lochblenden benutzt wird.
Auflösungsvermögen, optische Schnitte und Positionierungsgenauigkeit
Auflösung

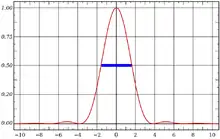
Wie generell bei Lichtmikroskopen ist die Auflösung auch bei konfokalen Mikroskopen durch Beugung begrenzt. Eine punktförmige Lichtquelle wird auf Grund der Beugung als dreidimensionale Punktspreizfunktion (engl. point spread function, PSF) abgebildet. Der Schnitt durch die mittlere Ebene der PSF wird als Beugungsscheibchen bezeichnet (siehe Abbildung). Aus praktischen Gründen wird in der Konfokalmikroskopie statt der Auflösung (Rayleigh-Kriterium) häufig die Halbwertsbreite (engl.: full width half maximum, FWHM) der PSF angegeben, also die Breite, bei der noch 50 Prozent der maximalen Helligkeit vorhanden sind. Diese Werte sind grundsätzlich etwas niedriger als die eigentliche Auflösung.
Durch die Lochblenden im Beleuchtungs- und im Detektionsstrahlengang kann die konfokale Auflösung etwas besser sein als in konventionellen Mikroskopen. Die größtmögliche theoretische Auflösungsverbesserung um den Faktor wird aber nur erreicht, wenn die Detektionslochblende nahezu völlig geschlossen ist, so dass dann kein Licht mehr aufgefangen und daher kein Bild entstehen würde.[21][22]

Die tatsächlich erzielbare Auflösung ist daher nur wenig besser als in konventionellen Mikroskopen. Liegt der Lochblendendurchmesser im ersten Minimum des Beugungsscheibchens (also im ersten schwarzen Ring), so ist die Auflösung in der Fokusebene nicht mehr besser als im nicht-konfokalen Fall, wogegen die Signalintensität dann schon fast maximal ist.[2] Dieser Wert ist in der Software von kommerziellen Konfokalmikroskopen häufig voreingestellt. Er wird als eine Airy Unit (AU) bezeichnet, nach den englischen Begriffen Airy disk (= Beugungsscheibchen) und unit (= Maßeinheit).
Wie generell bei Lichtmikroskopen ist die Auflösung in der Schärfeebene besser als entlang der optischen Achse (anisotrope Auflösung). In der Tabelle sind die Formeln zur Berechnung der Halbwertsbreite der Punktspreizfunktion angegeben.
| Ø > 1 AU | Ø < 0,25 AU | Objektiv NA=1,4/n=1,518 für Ø = 1 – 0,25 AU (λ=500 nm) | konfokales Volumen Isofläche gleicher Intensität | |
|---|---|---|---|---|
| FWHMlateral | 182 – 132 nm | 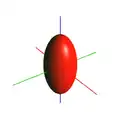 | ||
| FWHMaxial | 473 – 344 nm | |||
Formeln zur Berechnung der Halbwertsbreite (FWHM) der Punktspreizfunktion (PSF) bei einem Durchmesser Ø der Detektionslochblende von >1 AU (Spalte 2) und <0,25 AU (Spalte 3). NA: numerische Apertur des Objektivs; λ: Wellenlänge des Lichtes; n: Brechungsindex des Immersionsmediums (1 für Luft).[21][23] Spalte 3 enthält ein Beispiel für ein hochwertiges Ölimmersionsobjektiv, Spalte 4 eine Darstellung des Volumens, aus dem Signal am Detektor ankommt (optische Achse vertikal). | ||||




Die beschriebenen Zusammenhänge gelten nur für ideale optische Bedingungen. Eine dieser Bedingungen ist eine völlig gleichmäßige Ausleuchtung der hinteren Brennebene des Objektivs durch das Anregungslichts, da nur dann im Präparat ein ideales Beugungsscheibchen entsteht. Der Helligkeitsquerschnitt eines Laserstrahls zeigt jedoch ungefähr eine Gauß-Verteilung. Um den negativen Effekt zu minimieren, wird die rückwärtige Pupille des Objektivs überstrahlt, das heißt, der Laserstrahl wird so stark aufgeweitet, dass die äußeren Bereiche abgeschnitten werden und der verbleibende Querschnitt geringere Helligkeitsunterschiede hat. Wird so stark aufgeweitet, dass noch 50 % des Lichts durchtreten, so beträgt der Auflösungsverlust noch etwa 8 %; treten noch 30 % durch, liegt der Verlust bei 5 %.[23] Weiterhin kommt es bei der Untersuchung von Fluoreszenz zu einem Unterschied zwischen Anregungs- und Detektionswellenlänge (Stokes-Verschiebung), welche zu einem weiteren Auflösungsverlust führt. Die mittlere Wellenlänge ergibt sich näherungsweise zu .[23]

Ein weiterer Auflösungsverlust kann im Präparat verursacht werden, wenn der Brechungsindex des Einbettungsmediums oder die Deckglas-Dicke von den für das Objektiv vorgesehenen Werten abweicht und es dadurch zu sphärischen Aberrationen kommt.
Axiale Begrenzung des Signals und Positionierungsgenauigkeit
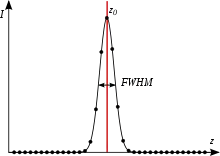
Die geringe Verbesserung der Auflösung rechtfertigt kaum den erhöhten Aufwand und die damit verbundenen Kosten. Der entscheidende Vorteil von Konfokalmikroskopen ist vielmehr die Möglichkeit, optische Schnitte aufzunehmen, denn bedingt durch die Lochblenden fällt die Intensität des Signals in etwa mit der vierten Potenz des Abstands zur Fokusebene ab (1/Abstand4).[24] Der Effekt lässt sich am Beispiel einer spiegelnden Oberfläche bei Reflexion erklären. In konventionellen Auflichtmikroskopen lässt sich die genaue Position der Oberfläche nicht feststellen, da die reflektierte Lichtmenge in den darüber und darunter liegenden Ebenen die gleiche ist. Im konfokalen Mikroskop wird jedoch nur Licht detektiert, wenn die spiegelnde Oberfläche im Bereich der Schärfeebene liegt. Dadurch ist die Genauigkeit einer Positionsbestimmung sehr hoch. Ebenso verbessert sich der Kontrast in fluoreszierenden Präparaten, da keine Fluoreszenz aus anderen Ebenen zum Detektor gelangt.[21]
Die Genauigkeit, mit der eine Position bestimmt werden kann, ist erheblich besser als die erzielbare Auflösung, da die Mitte eines Helligkeitsmaximums sehr genau festgestellt werden kann (siehe Abbildung). Dadurch kommt keine Unterschreitung der erzielbaren Auflösung zustande, da nicht feststellbar ist, ob das aufgenommene Signal von einer oder mehreren dicht beieinander liegenden Strukturen stammt. Diese Einschränkung ist unerheblich, wenn in der Materialforschung die Höhe von Oberflächen vermessen werden soll. Eine derartige Höhenvermessung ist also nicht durch die Auflösung begrenzt. Der beschränkende Faktor ist die Unsicherheit, mit der die Position der maximalen Intensität entlang der optischen Achse bestimmt werden kann. Die Unsicherheit ist in erster Linie durch das Systemrauschen beschränkt[25] und beträgt in einem gut aufgebauten Konfokalmikroskop bei Verwendung eines hochaperturigen Objektivs nur wenige Nanometer.
Verwandte Verfahren
Nicht-mikroskopische konfokale Techniken
Konfokale Techniken werden auch außerhalb der Mikroskopie eingesetzt, beispielsweise für Chromatisch-konfokale Abstandsmessungen. Eine Übersicht gibt der Artikel Konfokaltechnik.
In der Medizin wird konfokale Endomikroskopie als eine Methode der Endoskopie verwendet.
Andere Laser-Scanning-Mikroskope
- siehe auch: Laser-Scanning-Mikroskope
In gewisser Weise Vorläufer der konfokalen Laser-Scanning-Mikroskope sind Flying-Spot-Mikroskope. Bei Ihnen wird wie bei den konfokalen Punktscannern ein Beleuchtungspunkt über das Präparat geführt. Dieser ist aber nicht notwendigerweise beugungsbegrenzt. Auch fehlt eine Detektionslochblende. Der Beleuchtungspunkt wurde in frühen Geräten häufig durch eine Braun'sche Röhre erzeugt, nach der Entwicklung des Lasers auch durch einen fokussierten Laserstrahl. Derartige Geräte waren die ersten Laser-Scanning-Mikroskope.[26]
Andere Laser-Scanning-Mikroskope wurden dagegen erst nach der Entwicklung des konfokalen Laser-Scanning-Mikroskops gebaut. 4Pi-Mikroskope und STED-Mikroskope sind spezielle konfokale Mikroskopie, die eine verbesserte Auflösung ermöglichen. Multiphotonenmikroskopie ist dagegen ein nicht-konfokales Laser-Scanning-Verfahren, da keine Lochblenden mit konfokalen Fokuspunkten benötigt werden. Hier entsteht nur dann ein Signal, wenn zwei oder mehr Photonen gleichzeitig am Fokuspunkt eintreffen, daher der Name. Die drei genannten Verfahren sind häufig als Zusatzeinrichtung eines konfokalen Laser-Scanning-Mikroskops eingebaut.
Geschichte
1940: ein Spaltlampensystem zur Dokumentation von Augenuntersuchungen
Der Augenarzt Hans Goldmann, Direktor der Universitäts-Augenklinik im schweizerischen Bern, kämpfte mit dem Problem, dass Spaltlampen immer nur einen eng begrenzten Teil des Auges scharf abbildeten (Hornhaut oder Teil der Linse). Zwar konnte ein Beobachter ein durchgehendes Bild im Geist zusammensetzen, eine fotografische Darstellung beschränkte sich jedoch immer auf einen schmalen Bereich. Goldmann entwickelte ein Gerät zur „Spaltlampenphotographie und -photometrie“, das von der ortsansässigen Firma Haag-Streit hergestellt wurde.[27]
Der scharfe Anteil des Bildes wurde durch ein Objektiv auf einen Film projiziert. Durch eine spaltförmige Blende vor dem Film wurde verhindert, dass unscharfe Bildanteile abgelichtet wurden. Der Mechanismus zur Bewegung des Beleuchtungsspalts war über eine Scheibe fest mit der Filmtrommel verbunden: Bewegte sich der Spalt der scharfen Abbildung über das Auge, so drehte sich der Film in der Trommel entsprechend hinter der Spaltblende vorbei.[27]
Die Beleuchtung durch einen Spalt entspricht in etwa einer Lichtscheibe, die Beleuchtung ist also nicht auf eine Linie fokussiert, wie in späteren konfokalen Linienscannern. Die Beobachtungsachse in Goldmanns Apparat lag aber ungefähr im 45°-Winkel zur Beleuchtungsachse, so dass – vom Detektor aus gesehen – dennoch nur eine Linie (und nicht die Ebenen darunter oder darüber) beleuchtet wurde. Der Spalt vor der Kamera entsprach von der Funktion her der Schlitzblende vor dem Detektor eines konfokalen Linienscanners, sodass das System nachträglich als konfokal bezeichnet wurde.[28][29]
Goldmanns Arbeit wurde in den 1970er Jahren von D. M. Maurice zitiert, der einen konfokales Line-Scanning-Microscope für die Augenheilkunde entwickelte.[30] Auch historische Rückblicke erwähnen Goldmanns Apparat.[15][28][29]
1943, 1951: konfokale Mikroskope für die Spektrophotometrie
1943 veröffentlichte Zyun Koana eine Arbeit[31] über ein konfokales Mikro-Photometrie-System. Eine Abbildung zeigt das Schema eines konfokalen Transmissionsstrahlengangs: Die Beleuchtung gelangt durch zwei Linsen und die Anregungslochblende auf eine weitere Linse (entsprechend dem Kondensor in normalen Lichtmikroskopen), von der sie in das Präparat fokussiert wird. Das Licht durchtritt das Präparat und wird von einer weiteren Linse (entsprechend dem Objektiv) auf eine Detektionslochblende fokussiert. Von dieser japanischen Arbeit liegt keine Übersetzung oder Zusammenfassung in einer westlichen Sprachen vor.[28] Koana (1907–1985) wurde später bekannt für die Entwicklung von Fotoapparat-Objektiven in Zusammenarbeit mit der Firma Nikon.[32]
Hiroto Naora, ein Mitarbeiter Koanas, veröffentlichte 1951 eine Arbeit in der Zeitschrift Science. Er fokussierte eine Lochblende im Beleuchtungsstrahlengang 2000-fach verkleinert ins Präparat. Eine zweite Lochblende in der Zwischenbildebene minimierte Streulicht bei der Aufnahme. Ziel war es, kleine Bereiche in Zellkernen zu beleuchten, in welchen die DNA mit der Feulgenfärbung nachgewiesen wurde, um die DNA-Menge quantitativ zu bestimmen. Das Mikroskop erzeugte keine Bilder, stattdessen wurden die Intensität und die Wellenlängen des vom Präparat durchgelassenen Lichtes gemessen (Spektrophotometrie). Durch die beiden Lochblenden wurde Streulicht vermieden und so die Messgenauigkeit verbessert. Der erzeugte Beleuchtungsfleck hatte 1 – 5 µm Durchmesser und war also nicht beugungsbegrenzt. Auch wurde das Präparat nicht abgerastert.[33]
1955, 1957: der erste Punktscanner
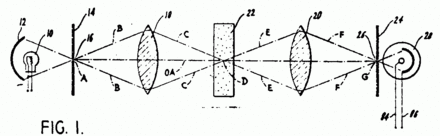
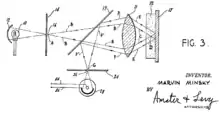
Das erste punkt-rasternde Konfokalmikroskop wurde von Marvin Minsky 1955 entwickelt und 1957 zum Patent[34] angemeldet. Er wollte Gehirnschnitte untersuchen und kämpfte mit starker Streuung in diesem dichten Gewebe. Da das helle Streulicht das Erkennen der eigentlichen Strukturen verhinderte, suchte er nach einer Möglichkeit dieses zu reduzieren und fand sie im konfokalen Prinzip.[35]
Das Mikroskop hatte einen waagrechten, unbeweglichen Strahlengang (siehe Schemazeichnung) und eine Zirconium-Bogenlampe als Lichtquelle. Die Öffnung der ersten Lochblende wurde durch ein 45x-Trockenobjektiv ins Präparat fokussiert. Das Präparat wurde zwischen zwei Deckgläser platziert und in eine bewegliche Vorrichtung eingespannt, die zum Rastern in x- und y-Richtung bewegt wurde („stage scanning“). Das zweite, gleichartige Objektiv nahm das durchgetretene Licht auf und leitete es durch die zweite Lochblende zum Detektor. Minsky setzte nie Ölimmersion ein, da er befürchtete, dass die Viskosität zu Problemen bei der Bewegung des Präparates führen könnte und dass die geringere Eindringtiefe der Ölimmersionsobjektive nicht ausreichend wäre. Das System konnte Punkte auflösen, die einen Abstand von weniger als einem Mikrometer hatten.[35]
Detektor war ein Photomultiplier, als Anzeigegerät diente ein Radarschirm (Kathodenstrahlröhre) mit einer Bildanzeigedauer von etwa zehn Sekunden. Dies war auch die Zeit, die das Rastern eines Bildes benötigte. Eine Digitalisierung oder fotografische Dokumentation war nicht eingebaut, Bilder sind nicht überliefert. Später führte Minsky den Mangel an Dokumentationsmöglichkeiten als einen Grund an, warum es noch 30 Jahre dauerte, bis sich die Idee des konfokalen Mikroskops durchsetzte.[35]
Minsky schrieb keine wissenschaftlichen Arbeiten, die das konfokale Mikroskop erwähnten. Sein Schwager, ein Patentanwalt, fand das Gerät interessant, und so kam es zur einzigen zeitgenössischen Dokumentation in einem Patentantrag. In diesem ist auch der Aufbau einer zweiten Version beschrieben, bei der Licht durch das Präparat hindurchgeht, danach auf einen Spiegel trifft und von diesem wieder zurückgeworfen wird (siehe Abbildung). Er erwähnte auch die Möglichkeit des Scannens entlang der optischen Achse (z-Richtung), die er aber ebenfalls nicht verwirklichte.[35]
1966: „Vorrichtung zur optischen Abtastung mikroskopischer Objekte“
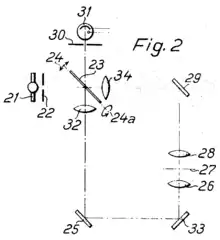 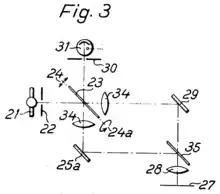 Weber'sche Varianten mit Kippspiegel, oben: Durchlicht. Von der Lichtquelle (21) geht es durch die Lochblende (22) auf den Kippspiegel (23), der in zwei Richtungen (24 und 24a) beweglich ist. Über Linsen und Spiegel geht es zum Präparat (27) und von dort über die verspiegelte Spiegel-Rückseite (23) und die Detektionslochblende (30) zum Detektor. In der Auflichtvariante (unten) sorgt ein halbdurchlässiger Spiegel (35) für Spiegelung zum Objektiv (28) und Präparat (27) und Durchlässigkeit zum Detektor (31). |
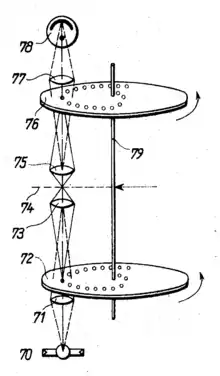 Weber’sche Variante mit Nipkow-Scheiben. Von der Lichtquelle (70) gelangt die Beleuchtung über eine Linse (71) zur unteren Nipkow-Scheibe. Nur durch ein Loch gelangt Licht zum Kondensor (73) um in der Schärfeebene (74) eine punktförmige Beleuchtung zu erzeugen. Vom Objektiv (75) wird das Licht von dieser Stelle über die zweite Nipkow-Scheibe zum photoelektrischen Empfänger (78) geleitet. |
Weniger bekannt ist die Entwicklung mehrerer Ansätze für konfokale Rastermikroskope durch Klaus Weber, Mitarbeiter der Ernst Leitz GmbH in Wetzlar.[36][37] Wie bei Koana und Naora war das Ziel nicht die Erstellung eines Bildes, sondern das Messen der Signalintensität in kleinen Ausschnitten des Präparats.
Für das Abrastern sah Weber drei alternative Möglichkeiten „zur synchronen Abtastung auf der Beleuchtungs- und der Beobachtungsseite“ vor:
- Eine klassische Nipkow-Scheibe (mit je nur einer Spirale aus Lochblenden) kam in die Ebene der Leuchtfeldblende und eine zweite, synchron laufende in die Zwischenbildebene (siehe linke Abbildung).
- In die Leuchtfeld- und die Zwischenbildebene kam je eine bewegliche Lochblende, die in einer Richtung (x-Achse) synchron elektrisch bewegt wurden. Das Rastern entlang der y-Achse geschah durch Bewegung des Objekts oder ebenfalls durch Bewegung der Lochblende.
- Diese „besonders vorteilhafte Anordnung“ verwendete einen Kippspiegel, der das Bild der Beleuchtungslochblende („Leuchtfleckblende“) als „Leuchtfleck“ über das Präparat bewegte, und über den auch der Beobachtungsstrahlengang zur Detektionslochblende („Bildfeldblende“) lief. Das Präparat war also stationär und wurde durch Verschiebung des Strahlengangs abgerastert (beam scanning).[36][38]
Von der Kippspiegelvariante schlug Walter wiederum mehrere Ausführungen vor. Dabei sollte entweder Beleuchtungs- und Beobachtungslicht über die gleiche Seite des Spiegels laufen, oder, „besonders zweckmäßig“, der Beobachtungsstrahlengang lief über die ebenfalls verspiegelte Rückseite, so dass Beleuchtungs- und Beobachtungsstrahlengang zwar getrennt, aber dennoch synchronisiert waren, so dass der im Präparat erzeugte Lichtfleck immer auf die Detektionslochblende abgebildet wurde. Für diese Lösung zeigt der Patentantrag eine Version für Durchlicht und eine für Auflicht-Reflexionsmikroskopie (siehe Abbildungen). Die Möglichkeit das elektrische Signal des Detektors zur Erstellung eines Bildes zu nutzen wie beim Minsky-Mikroskop wurde in den Patenten nicht erwähnt.[36]
Es ist unklar, ob die von Weber vorgeschlagenen Geräte tatsächlich gebaut wurden und ob seine Ideen Einfluss auf weitere Entwicklungen hatten. Das erste Patent, das Webers US-Patent zitierte, wurde 1980 eingereicht.[37]
Weber hatte nicht als Erster die Idee, eine Nipkow-Scheibe in Mikroskopen einzusetzen. Bereits 1951 wurde ein System vorgestellt, bei dem eine klassische Nipkow-Scheibe eingesetzt wurde, um ein Fluoreszenzbild so zu zerlegen, dass die Helligkeit der einzelnen Bildpunkte nacheinander von einem Photomultiplier gemessen werden konnte. Es handelte sich also nicht um ein konfokales Abbildungssystem, sondern um einen „Microfluorometric Scanner“.[39][28]
1967: das erste bildgebende konfokale Mikroskop mit Nipkow-Scheibe
In den 1960er Jahren entwickelte der Tschechoslowake Mojmír Petráň von der Medizinischen Fakultät der Karls-Universität in Pilsen, das oben beschriebene Tandem-Scanning-Mikroskop. Es war das erste konfokale Mikroskop, das zum Verkauf angeboten wurde: zum einen von einer kleinen Firma in der Tschechoslowakei und zum anderen in den USA von Tracor-Northern (später Noran).[13]
Der ausgebildete Arzt Petráň besuchte 1964 die Arbeitsgruppe von Robert Galambos an der Yale University in New Haven (Connecticut, USA). Sie überlegten, wie unfixierte, ungefärbte Nervenzellen im Gehirn beobachtet werden könnten und entwickelten während dieses Aufenthaltes das Konzept. Im folgenden Jahr bauten Petráň und Milan Hadravský in Pilsen den ersten Prototyp.[40]
Das tschechoslowakische Patent wurde 1966 von Petráň und Hadravský eingereicht. 1967 erschien eine erste wissenschaftliche Veröffentlichung in der Zeitschrift Science, welche mit dem Mikroskop gewonnene Daten und Abbildungen enthielt. Autoren waren M. David Egger von der Yale University und Petráň.[16] In den Fußnoten dieser Arbeit heißt es, dass Petráň das Mikroskop entworfen und seine Konstruktion geleitet hatte und dass er zeitweise ein „research associate“ an der Yale University war. 1968 erschien eine weitere Arbeit, in der zusätzlich Hadravský und Galambos Autoren waren.[18] Hier wurden Theorie und technische Details des Mikroskops beschrieben. 1970 wurde das 1967 beantragte US-Patent erteilt.[17] Es enthält auch eine Version des Mikroskops für Durchlicht: Das Präparat wird durch ein Objektiv beleuchtet und durch ein weiteres, identisch gebautes, beobachtet. Es ist jedoch unklar, ob diese Variante tatsächlich gebaut wurde.
Beim Jahrestreffen der European Light Microscopy Initiative (ELMI) 2011 wurden Petráň und Hadravský für Ihre Verdienste geehrt,[41] und 2012 wurden beide vom Projekt zur Kulturhauptstadt Europas „Plzeň 2015“, in ihrer Heimatstadt zu „Pilsener Ikonen“ ernannt. Für Hadravský wurde diese Ehrung in memoriam verliehen.[42]
Erst 1986 wurde das Tandem-Scanning-Mikroskop zur Untersuchung des Auges verwendet. Nach erfolgreichen ex vivo Versuchen entwickelten Wissenschaftler an der Georgetown University in den USA das Gerät so weiter, dass es auch an Patienten eingesetzt werden konnte. Eine kommerzielle Version wurde von Tandem Scanning Corporation, Inc. entwickelt. Das Gerät enthielt eine zusätzliche Linse, deren Bewegung die Schärfeebene im Auge veränderte, ohne dass das Objektiv bewegt werden musste.[15]
1969: das erste konfokale Laser-Scanning-Mikroskop
Egger war an einer weiteren Entwicklung beteiligt. Zusammen mit Paul Davidovits, ebenfalls Yale University, veröffentlichte er 1969[43] und 1971[8] zwei Arbeiten über das erste konfokale Mikroskop, das mit Laserlicht arbeitete, einen Punktscanner. Auch dieses Gerät war für Auflicht-Reflexionsmikroskopie vorgesehen, im Besonderen für die Beobachtung von Nervengewebe. Bereits 1969 spekulierten die Autoren über einen Einsatz von Fluoreszenzfarbstoffen für "in vivo"-Untersuchungen. Sie zitierten das Patent von Minsky und bedankten sich bei Steve Baer für den Vorschlag, einen Laser mit ‚Minskys Mikroskop‘ zu verwenden, sowie bei Galambos, Hadravsky und Petráň für Diskussionen, die zur Entwicklung des Mikroskops führten. Baer war Doktorand an der Albert Einstein School of Medicine in New York City, wo er ein konfokales Line-Scanning-Mikroskop entwickelte.[44] Als Motivation für die Neuentwicklung gaben Egger und Davidovits in der zweiten Arbeit an, dass im Tandem-Scanning-Mikroskop nur ein Anteil von 10−7 der Beleuchtung im Okular zur Bildentstehung beitrage und die Bildqualität daher für die meisten biologischen Untersuchungen nicht ausreiche.[45][28][40]
Ein Helium-Neon-Laser mit 633 nm Wellenlänge und 5 mW Leistung wurde auf einen halbdurchlässigen Spiegel geleitet und von diesem zum Objektiv reflektiert. Als Objektiv diente eine einfache Linse mit einer Brennweite von 8,5 mm. Im Gegensatz zu allen früheren und den meisten späteren Entwicklungen wurde das Präparat durch Bewegen dieser Linse abgerastert (objective scanning), wodurch sich der Fokuspunkt entsprechend verschob. Reflektiertes Licht gelang zurück zum halbdurchlässigen Spiegel, und der durchgelassene Anteil wurde von einer weiteren Linse zur Detektionslochblende geleitet, hinter der sich ein Photomultiplier befand. Das Signal wurde von der Kathodenstrahlröhre eines Oszilloskops angezeigt, wobei der Kathodenstrahl synchron mit dem Objektiv bewegt wurde. Eine spezielle Apparatur konnte Polaroid-Fotos von der Anzeige machen, von denen drei in der Veröffentlichung von 1971 wiedergegeben sind. Laserlicht ist grundsätzlich linear polarisiert. In der ersten der beiden Veröffentlichungen bewirkte ein Analysator (ein Polarisationsfilter) vor der Detektionslochblende, dass Licht, welches innerhalb des Mikroskops reflektiert wurde, nicht zum Detektor gelangen konnte. Ein λ/4-Plättchen zwischen Objektiv und Präparat sorgte dafür, dass das vom Präparat reflektierte Licht um insgesamt 90° gegenüber der Laserpolarisation verschoben wurde, so dass dieses durch den Analysator gelang. In der Mikroskopversion der zweiten Veröffentlichung fehlten diese beiden Bauteile jedoch.[43][8]
Davidovits, Egger und Marvin Minsky erhielten 2000 den R. W. Wood Prize der Optical Society of America für Beiträge zur Entwicklung des Konfokalmikroskops.
1977–1985: Punktscanner mit Laser und Rasterung durch Präparatbewegung (stage scanning)
Colin J. R. Sheppard und A. Choudhury in Oxford veröffentlichten 1977 eine theoretische Analyse von Konfokalmikroskopie und Laser-Scanning-Mikroskopie. Diese Arbeit[46] ist vermutlich die erste Veröffentlichung, die den Ausdruck „confocal microscope“ enthält.[45][28]
Die Brüder Christoph Cremer und Thomas Cremer in Heidelberg entwarfen 1978 ein konfokales Laser-Scanning-Mikroskop für die Fluoreszenzanregung mit elektronischem Autofokus. Sie schrieben: „Aufgrund seiner besonderen Darstellungsmöglichkeiten könnte das Laser-Scanning-Mikroskop-Verfahren eine wertvolle Ergänzung herkömmlicher lichtmikroskopischer sowie rasterelektronenmikroskopischer Verfahren werden.“ Sie schlugen auch ein Laser-Punkt-Beleuchtung mit Hilfe eines „4π-Punkt-Hologramms“ vor.[45][47]
Die Oxford-Gruppe um Sheppard und Tony Wilson beschrieb 1978 und 1980 ein Auflicht-Konfokal-Mikroskop mit Stage-scanning, Laserbeleuchtung und Photomultipliern als Detektoren. Das Präparat konnte nicht nur in der Fokusebene, sondern auch entlang der optischen Achse bewegt werden, wodurch die Aufnahme von optischen Serienschnitten möglich wurde. Die Vorzüge des Gerätes konnten besonders überzeugend an integrierten elektronischen Schaltkreisen gezeigt werden.[45] Der Begriff und die Methodes des „optical sectioning“ ist allerdings schon älter. Bereits 1930[48] zeigte Francis Lucas optische Serienschnitte, die er mit einem UV-Licht-Mikroskop auf Film erzeugte.[28]
Fred Brakenhoff und Mitarbeiter wiesen 1979 nach, dass die theoretischen Vorteile der optischen Schnitte und der Auflösungsverbesserung tatsächlich erreichbar sind. 1985 veröffentlichte die Gruppe die ersten überzeugenden Bilder zu zellbiologische Fragestellungen, die sie mit einem weiterentwickelten konfokalen Mikroskop aufnahmen. Weitere biologische Anwendungen folgten kurz darauf von anderen Gruppen.[24]
Zwischenzeitlich veröffentlichten I. J. Cox und Sheppard aus der Oxford-Gruppe 1983 die erste Arbeit[49] über ein konfokales Mikroskop, das mit einem Computer verbunden wurde.[40]
Das erste kommerzielle konfokale Laser-Scanning-Mikroskop, der Stage-Scanner SOM-25, wurde ab 1982 von Oxford Optoelectronics (über Zwischenschritte von BioRad übernommen) angeboten; es basierte auf dem Design der Oxford-Gruppe.[45][28]
Ab 1985: Laser-Punktscanner mit Beam-Scanning
Die meisten bisher entwickelten konfokalen Laser-Scanning-Mikroskope gehörten zum Stage-Scanning-Typ: Der Beleuchtungspunkt war unbeweglich und der Objekttisch (englisch stage) mit dem Präparat wurde in x- und y-Richtung bewegt, um die Objekte in der Fokusebene abzurastern. Dieses Verfahren war langsam und empfindlich bezüglich Erschütterungen. Bereits Minsky hatte die Möglichkeit erwähnt, stattdessen einen rasternden Lichtstrahl über das unbewegliche Präparat zu schwenken, dies jedoch wegen technischen Schwierigkeiten verworfen.[24][40]
Mitte der 1980er Jahre entwickelten W. B. Amos, J. G. White und Mitarbeiter in Cambridge das erste konfokale Beam-Scanning Mikroskop (beam, engl. für Strahl, Lichtstrahl), bei dem das Präparat still stand und stattdessen der Beleuchtungspunkt bewegt wurde. Dadurch konnten vier Bilder pro Sekunde mit jeweils 512 Zeilen aufgenommen werden. Die Idee des Beam-Scannings wurde aus Flying-Spot-Mikroskopen übernommen. Eine zweite wichtige Neuentwicklung war eine stark vergrößerte Zwischenbildebene durch einen um ein bis zwei Meter verlängerten Strahlengang. Das erzeugte Zwischenbild war dadurch 80-mal größer als durch die Objektivvergrößerung, mit der Folge, dass für die Lochblende eine gewöhnliche Irisblende mit einem Durchmesser von etwa einem Millimeter eingesetzt werden konnte, im Gegensatz zu den nur wenige Dutzend Mikrometer großen Lochblenden in früheren Systemen. Dadurch war die Justierung erheblich einfacher.[24]
Erste Fotografien wurden per Langzeitbeleuchtung mit Film gemacht, bevor eine digitale Kamera eingebaut wurde. Bilder von verschiedenen biologischen Präparaten waren, verglichen mit normaler Fluoreszenzmikroskopie, deutlich besser. Eine erste Veröffentlichung solcher Bilder erfolgte 1987. Eine weitere Geräteverbesserung erlaubte erstmals das hineinzoomen in das Präparat, also die Auswahl eines Teilbereichs für eine vergrößerte Darstellung oder eine schnellere Aufnahme. Zeiss, Leitz und Cambridge Instruments hatten an einer kommerziellen Produktion kein Interesse. Das Medical Research Council (MRC) erklärte sich jedoch bereit, die Entwicklung eines kompakteren Prototyps zu finanzieren. Schließlich übernahm Bio-Rad das Design, und eine Software für die Computersteuerung wurde entwickelt. Das Gerät kam als MRC 500 auf den Markt, der Nachfolger hieß MRC 600. Dieses Gerät war auch Grundlage für das an der US-amerikanischen Cornell University entwickelte und 1990 publizierte erste Zwei-Photonen-Fluoreszenzmikroskop.[24]
Parallel gab es eine weitere Entwicklung an der Universität Stockholm, die etwa gleichzeitig zu einem kommerziellen Gerät führte, das von der schwedischen Firma Sarastro vertrieben wurde. Diese wurde 1990 von Molecular Dynamics, einer US-amerikanischen Firma übernommen,[50] aber schließlich wurde die Produktion eingestellt. In Deutschland entwickelte die 1984 gegründete Firma Heidelberg Instruments ein konfokales Laserscanningmikroskop, das weniger für biomedizinische als für Industrieanwendungen entwickelt wurde.[51] Dieses Gerät wurde 1990 von Leica Lasertechnik übernommen und weiterentwickelt. Zeiss hatte bereits ein nicht-konfokales Flying-Spot Laserscanning-Mikroskop auf dem Markt, das zu einem konfokalen Mikroskop erweitert wurde. Ein Bericht von 1990,[52] der „einige“ Hersteller erwähnt, zählt die folgenden auf: Sarastro, Technical Instrument, Meridian Instruments, Bio-Rad, Leica, Tracor-Northern und Zeiss.[24]
Neuentwicklungen mit Nipkow-Scheibe
Die vielen Spiegel und Prismen des Tandem-Scanners, die Anregungslochblenden und Detektionslochblenden im Strahlengang genau aufeinander abstimmen und zu justieren, ist schwierig; auch das Flattern der dünnen Nipkow-Scheibe führt zu Justierschwierigkeiten. Schon Egger und Petráň schlugen daher vor, Anregungs- und Detektionsstrahlengang durch dieselben Lochblenden zu führen („einseitige Nipkow-Scheibe“). Sie verwarfen diesen Ansatz jedoch, da die Reflexion des Anregungslichts an der Oberfläche der Scheibe nicht vom Detektionsstrahlengang zu trennen war. Albert Frosch und Hans Erdmann Korth bekamen 1975 ein US-Patent[53] für IBM zugesprochen, in dem dieses Problem durch Schrägstellen der Nipkow-Scheibe angegangen wurde.[40]
Diese Idee wurde mit weiteren Verbesserungen die Grundlage für ein 1988 von Gordon S. Kino und Guoqing Xiao, Stanford University eingereichtes Patent über ein einseitiges Nipkow-Scheiben-Mikroskop für Auflicht-Reflexionsmikroskopie.[54] Das Gerät war für die Vermessung von Halbleitern gedacht. Vorteile waren eine geringere Vibrationsanfälligkeit und eine vereinfachte Justierung. Außerdem konnte die Nipkow-Scheibe nun quer zum Strahlengang bewegt werden, so dass in unterschiedlichen Spuren zwischen Scheibenmittelpunkt und Rand unterschiedlich große Lochblenden angebracht werden konnten. Damit konnte die Lochblendengröße an die Auflösung verschiedener Objektive angepasst werden, oder an die unterschiedlich starke Reflexion von verschiedenen Präparateregionen.[40][55]
Zur Verminderung der Reflexion direkt von der Nipkow-Scheibe zum Detektor wurde eine geschwärzte, um 5° zur optischen Achse geneigte Scheibe eingebaut. Nach der Lichtquelle kamen ein Polarisator und dahinter ein halbdurchlässiger Spiegel, der zur Nipkow-Scheibe reflektierte. Zwischen Scheibe und Objektiv war ein Lambda-Viertel-Plättchen, auf dem Rückweg des Lichts, direkt vor dem Detektor, war ein Analysator. Zwar ging so ein größerer Teil des Lichts verloren, das direkt von der Nipkow-Scheibe reflektierte Licht wurde jedoch effektiv vom Analysator blockiert.[40]
Ichihara und Kollegen veröffentlichen 1996 die erste Arbeit zum Yokogawa-Spinning-Disk-System (siehe oben).[45]
Weblinks
- Optical Microscopy Primer (englisch) Umfangreiche Linksammlung zu detaillierten Beschreibungen der Mikroskopie u. a. auch virtuelle Konfokalmikroskope
- Die konfokale Laser Scanning Mikroskopie Einführung in die konfokale Mikroskopie von Zeiss (PDF-Datei, 884 KB)
Literatur
Weiterführende Literatur
- Michael Volger: Lichtmikroskopie - Theorie und Anwendung. Irene K. Lichtscheidl, Universität Wien (Hrsg.), Onlineausgabe 29. Februar 2008, PDF-Datei auf: univie.ac.at. (Abhandlung über Lichtmikroskopie, S. 174–220 zur Fluoreszenz- und Konfokalmikroskopie.)
- Michiel Müller: Introduction to Confocal Fluorescence Microscopy (Tutorial Texts in Optical Engineering). 2. Auflage. SPIE Press, 2006, ISBN 0-8194-6043-5. Verlagswebsite (Einführung in die konfokale Fluoreszenzmikroskopie.)
- Guy Cox: Optical Imaging Techniques in Cell Biology. 1. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2006, ISBN 0-8493-3919-7. (2. Auflage. 2012, ISBN 978-1-4398-4825-8) (Allgemeine Einführung in die Lichtmikroskopie, mit Kapiteln speziell zur konfokalen Mikroskopie sowie digitalen Bildern, Aberrationen, Fluoreszenz etc.)
- James Pawley (Hrsg.): Handbook of Biological Confocal Microscopy. 3. Auflage. Springer Science and Business Media, 2006, ISBN 0-387-25921-X. (Das Nachschlagewerk zur konfokalen Mikroskopie in den Lebenswissenschaften, weniger für Einsteiger gedacht.)
Einzelnachweise
- Guy Cox: Optical Imaging Techniques in Cell Biology. 1. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2006, ISBN 0-8493-3919-7, S. 57–75.
- Colin J. R. Sheppard, David M. Shotton: Confocal Laser Scanning Microscopy. In: Royal Microscopical Society Microscopy Handbooks. Band 38. BIOS Scientific Publishers Limited, Oxford, UK 1997, ISBN 1-872748-72-4, S. 37, 39–42.
- Hybriddektor R10467U-40 (Seite nicht mehr abrufbar, Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis. auf der Website von Hamamatsu Photonics K.K., abgerufen am 4. Dezember 2012.
- Leica HyD for Confocal Imaging. (PDF; 1,9MB) Leica Microsystems, abgerufen am 9. Februar 2016.
- The HPM-100-40 Hybrid Detector. (PDF; 1,4MB) Becker & Hickl GmbH, abgerufen am 9. Februar 2016.
- PMA Hybrid Series | PicoQuant. PicoQuant GmbH, abgerufen am 14. Juli 2017.
- Konfokale Mikroskope mit Stage Scanning werden von mehreren Herstellern angeboten (z. B. MicroTime von PicoQuant, DeltaMyc von Horiba).
- P. Davidovits, M. D. Egger: Scanning laser microscope for biological investigations. In: Applied optics. Band 10, Nummer 7, Juli 1971, S. 1615–1619, ISSN 0003-6935. PMID 20111173. doi:10.1364/AO.10.001615
- MicroTime von PicoQuant, Website des Herstellers, abgerufen am 14. Juli 2017.
- DeltaMyc von Horiba, Website des Herstellers, abgerufen am 14. Juli 2017.
- Alba FCS, Website des Herstellers, abgerufen am 26. Januar 2012.
- konfokales Ramanmikroskop von Witec (Memento vom 22. Dezember 2012 im Internet Archive) Website des Herstellers, abgerufen am 26. Januar 2012.
- Guy Cox: Optical Imaging Techniques in Cell Biology. 1. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2006, ISBN 0-8493-3919-7, S. 115–122.
- Website von Zeiss zum Axio CSM 700 Echtfarben-Konfokalmikroskop, dort auch als PDF-Datei erhältlich, abgerufen am 26. Januar 2012.
- J. C. Erie: Corneal wound healing after photorefractive keratectomy: a 3-year confocal microscopy study. In: Transactions of the American Ophthalmological Society. Band 101, 2003, S. 293–333, ISSN 0065-9533. PMID 14971584. PMC 1358995 (freier Volltext).
- Egger MD, Petrăn M: New reflected-light microscope for viewing unstained brain and ganglion cells. In: Science. Band 157, Nr. 786, Juli 1967, S. 305–7, doi:10.1126/science.157.3786.305, PMID 6030094.
- Mojmír Petráň, Milan Hadravský: Method and arrangement for improving the resolving power and contrast. online bei Google Patents, Eingereicht 4. November 1967, erteilt 30. Juni 1970.
- Mojmír Petráň, Milan Hadravský, M. David Egger, Robert Galambos: Tandem-Scanning Reflected-Light Microscope. In: Journal of the Optical Society of America. Band 58, Nr. 5, 1968, S. 661–664, doi:10.1364/JOSA.58.000661.
- Heike Schmidt, Jürgen Valentin: Konfokal messen - 3D Oberflächenmessung per Mikroskop. In: Praxis Profiline. November 2006. PDF-Datei
- Andromeda iMIC – High-speed Live Cell Imaging with Confocal Resolution. Broschüre von Till Photonics. Online verfügbar als PDF-Datei (Memento vom 16. August 2012 im Internet Archive)
- B. Amos, G. McConnell, T. Wilson: Confocal microscopy. In: E. Egelman (Hrsg.): Comprehensive Biophysics. Elsevier, Amsterdam 2012.
- G. Cox, C. J. Sheppard: Practical limits of resolution in confocal and non-linear microscopy. In: Microscopy research and technique. Band 63, Nummer 1, Januar 2004, S. 18–22, ISSN 1059-910X. doi:10.1002/jemt.10423. PMID 14677129.
- Stefan Wilhelm, Bernhard Gröbler, Martin Gluch, Hartmut Heinz: Die konfokale Laser Scanning Mikroskopie. 09/03 Auflage. Carl Zeiss Mikroskopsysteme, Jena 2003 (PDF; 884kB).
- W. B. Amos, J. G. White: How the confocal laser scanning microscope entered biological research. In: Biology of the cell / under the auspices of the European Cell Biology Organization. Band 95, Nummer 6, September 2003, S. 335–342, ISSN 0248-4900. PMID 14519550. (Review).
- VDI/VDE 2655-1.2: Optische Messtechnik an Mikrotopografien; Kalibrieren von konfokalen Mikroskopen und Tiefeneinstellnormalen für die Rauheitsmessung. Inhalt-Entwurf (PDF; 112 kB)
- M. Françon: Einführung in die neueren Methoden der Lichtmikroskopie. Verlag G. Braun, Karlsruhe 1967, S. 253–256.
- Hans Goldmann: Spaltlampenphotographie und -photometrie. In: Ophthalmologica. Band 98, Nr. 5/6, 1939, S. 257–270, doi:10.1159/000299716. Hinweis: Der Band 98 wird zwar dem Jahr 1939 zugerechnet, auf der ersten Seite des Artikels ist als Erscheinungsdatum jedoch Januar 1940 angegeben.
- Colin JR Sheppard: Confocal Microscopy. The Development of a Modern Microscopy. In: Imaging & Microscopy. 3. November 2009 (online [abgerufen am 17. November 2012]).
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy. The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington, USA 2006, ISBN 978-0-8194-6118-6, S. 120–121.
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy. The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington, USA 2006, ISBN 978-0-8194-6118-6, S. 126.
- Zyun Koana: 微小部濃度計に關する諸問題. In: Journal of the Illumination Engineering Institute. Band 26, Nr. 8, 1942, S. 371–385. Die Arbeit (online PDF-Datei, 19020 Kilobyte) Abbildung 1b der Arbeit (S. 373) zeigt das Schema eines konfokalen Transmissionsstrahlengangs
- Dr. Zyun Koana Special Exhibition at the University of Tokyo (Memento vom 2. April 2015 im Internet Archive), Website von Michio Akiyama, aufgerufen am 8. Januar 2013.
- Hiroto Naora: Microspectrophotometry and cytochemical analysis of nucleic acids. In: Science. Band 114, Nummer 2959, September 1951, S. 279–280, ISSN 0036-8075. PMID 14866220. doi:10.1126/science.114.2959.279
- Marvin Minsky: Microscopy Apparatus. US Patent 3.013.467, Eingereicht 7. November 1957, erteilt 19. Dezember 1961.
- Marvin Minsky: Memoir on inventing the confocal scanning microscope. In: Scanning. Band 10, Nr. 4, 1988, S. 128–138, doi:10.1002/sca.4950100403.
- Patent DE1472293A1: Vorrichtung zur optischen Abtastung mikroskopischer Objekte. Angemeldet am 10. August 1966, veröffentlicht am 23. Januar 1969, Anmelder: Ernst Leitz G.m.b.H., Erfinder: Klaus Weber.
- Patent US3518014: Device for optically scanning the object in a microscope. Angemeldet am 7. August 1967, veröffentlicht am 30. Juni 1970, Anmelder: Ernst Leitz G.m.b.H., Erfinder: Klaus Weber.
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy. The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington, USA 2006, ISBN 978-0-8194-6118-6, S. 78–79.
- R. C. MELLORS, R. SILVER: A micro-fluorometric scanner for the differential detection of cells; application of exfoliative cytology. In: Science. Band 114, Nummer 2962, Oktober 1951, S. 356–360, ISSN 0036-8075. PMID 14883859.
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy. The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington, USA 2006, ISBN 978-0-8194-6118-6, S. 88–96.
- 11th annual meeting of the European Light Microscopy Initiative (ELMI), 2011, Meeting's Programme (Memento vom 31. August 2013 im Internet Archive)
- Plzeň 2015 kürt Pilsner Ikonen 2012. In: bbkult.net. Centrum Bavaria Bohemia, abgerufen am 9. Februar 2016.
- P. Davidovits, M. D. Egger: Scanning laser microscope. In: Nature. Band 223, Nummer 5208, August 1969, S. 831, ISSN 0028-0836. PMID 5799022. doi:10.1038/223831a0
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy. The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington, USA 2006, ISBN 978-0-8194-6118-6, S. 124–125.
- Shinya Inoué: Foundations of Confocal Scanned Imaging in Light Microscopy. In: James Pawley (Hrsg.): Handbook of Biological Confocal Microscopy. 3. Auflage. Springer Science and Business Media LLC, 2006, ISBN 978-0-387-25921-5, Kapitel 1, S. 1–19.
- C.J.R. Sheppard, A. Choudhury: Image Formation in the Scanning Microscope. In: Optica Acta: International Journal of Optics. 24, 1977, S. 1051–1073, doi:10.1080/713819421.
- C. Cremer, T. Cremer: Considerations on a laser-scanning-microscope with high resolution and depth of field. In: Microscopica acta. Band 81, Nummer 1, September 1978, S. 31–44, ISSN 0044-376X. PMID 713859.
- F. F. Lucas: THE ARCHITECTURE OF LIVING CELLS-RECENT ADVANCES IN METHODS OF BIOLOGICAL RESEARCH-OPTICAL SECTIONING WITH THE ULTRA-VIOLET MICROSCOPE. In: Proceedings of the National Academy of Sciences. Band 16, Nummer 9, September 1930, S. 599–607, ISSN 0027-8424. PMID 16587612. PMC 526698 (freier Volltext).
- I. J. Cox, C. J. Sheppard: Scanning optical microscope incorporating a digital framestore and microcomputer. In: Applied optics. Band 22, Nummer 10, Mai 1983, S. 1474, ISSN 0003-6935. PMID 18195988.
- Brent Johnson: Image Is Everything. In: The Scientist. 1. Februar 1999 (online [abgerufen am 30. November 2012]).
- Website von Heidelberg Instruments GmbH. Abgerufen am 4. Juni 2014.
- Diana Morgan: Confocal Microscopes Widen Cell Biology Career Horizons. In: The Scientist. 23. Juli 1990 (online [abgerufen am 30. November 2012]).
- Albert Frosch, Hans Erdmann Korth: Method of increasing the depth of focus. US Patent 3,926,500. bei Google Patents
- Gordon S. Kino, Guoqing Xiao: Scanning confocal optical microscope including an angled apertured rotating disk placed between a pinhole and an objective lens. US Patent 4927254, beantragt am 29. Juli 1988, erteilt am 22. Mai 1990 online bei Google Patents
- Shinya Inoué: Foundations of Confocal Scanned Imaging in Light Microscopy. In: James Pawley (Hrsg.): Handbook of Biological Confocal Microscopy. 3. Auflage. Springer Science and Business Media LLC, 2006, ISBN 0-387-25921-X, Kapitel 1, S. 1–19.