Lichtscheibenmikroskopie
Die Lichtscheibenmikroskopie bzw. Lichtscheibenfluoreszenzmikroskopie (LSFM, von englisch Lightsheet Fluorescence Microscopy, auch SPIM, von englisch Single Plane Illumination Microscopy, Selective Plane Illumination Microscopy, auch Lightsheet Microscopy und Lichtblattmikroskopie) ist ein fluoreszenzmikroskopisches Verfahren, bei dem nur eine dünne Schicht in der Probe beleuchtet wird, typischerweise einige Mikrometer. Verglichen mit herkömmlicher Fluoreszenzmikroskopie führt dies zu besserer Auflösung und deutlich vermindertem Bildhintergrund. Außerdem werden negative Effekte durch Bleichen oder lichtinduzierten Stress in biologischen Proben vermindert.
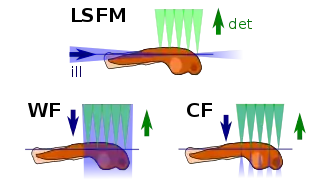

Das Verfahren wird in der Zellbiologie[1] und auch zu Fluoreszenzuntersuchungen an lebenden Organismen verwendet.[2] Viele Anwendungen finden sich auch bei Langzeitbeobachtungen der Embryonalentwicklung in Modellorganismen (Entwicklungsbiologie).
Die Anfang des 21. Jahrhunderts entwickelte Lichtscheibenmikroskopie[3][4] führte eine Beleuchtungsgeometrie in die Fluoreszenzmikroskopie ein, die in vergleichbarer Form Anfang des 20. Jahrhunderts mit dem Spaltultramikroskop bereits erfolgreich in der Dunkelfeldmikroskopie verwendet wurde.[5]
Aufbau
Grundlegender Aufbau
Bei dieser Art der Mikroskopie[6] wird senkrecht zur Beobachtungsrichtung Anregungslicht eingestrahlt (typischerweise durch einen Laser, der auf die Absorptionsbanden des gewählten Fluoreszenzfarbstoffes abgestimmt ist, z. B. aus einem Argon-Laser bei 488 nm für grün fluoreszierendes Protein). Der aufgeweitete, kollimierte Laserstrahl wird mit Hilfe einer Zylinderlinse nur in einer Richtung fokussiert. So ergibt sich im Fokus eine „Lichtscheibe“, die nur eine dünne Schicht innerhalb der Probe ausleuchtet. Um die numerische Apertur der Lichtscheibe zu erhöhen (und ihre Dicke so zu reduzieren), wird üblicherweise eine Kombination aus Zylinderlinse und einem Mikroskopobjektiv eingesetzt. Fluoreszenzfarbstoffmoleküle in der ausgeleuchteten Schicht werden zur Fluoreszenz angeregt, welche dann senkrecht dazu mit Hilfe eines Lichtmikroskops beobachtet wird. Um genug Platz für die Projektion der Lichtscheibe zu haben, werden üblicherweise sog. Tauchobjektive mit großem Arbeitsabstand (z. B. 2–3 mm bei einer numerischen Apertur von 1) eingesetzt, die vollständig in Wasser bzw. in eine Pufferlösung eintauchen. Daher wird in den meisten SPI-Mikroskopen um die Probe eine wassergefüllte Probenkammer konstruiert, die es auch erlaubt, die Probe bei physiologischen Bedingungen zu untersuchen (z. B. physiologische Salzkonzentrationen und 37 °C).
Das Fokussieren unterschiedlicher Teile der Probe erfolgt hier (im Gegensatz zur Weitfeld-Fluoreszenzmikroskopie) typischerweise nicht durch Verschieben des Objektives (dann müsste auch die Position der Lichtscheibe entsprechend geändert werden), sondern durch das Verschieben der Probe selbst.
Einige Erweiterungen der Technik
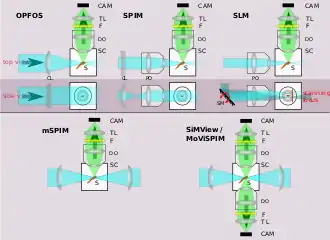
Seit den ersten Implementierungen des SPIM-Prinzips, wurden einige Erweiterungen vorgestellt, die die Eigenschaften eines SPI-Mikroskops verbessern oder den Aufbau vereinfachen:
- Zwei gegenläufige Lichtscheiben verringern typische SPIM-Artefakte, wie z. B. Abschattungen (siehe erster z-stack oben).[7]
- Zusätzlich zu den gegenläufigen Lichtscheiben wurde 2012 vorgeschlagen zwei Detektionsarme in ein SPIM zu integrieren, was die Messung von z- und Rotations-Stacks deutlich verschnellert.[8][9] Beide zusammen sind für eine vollständige 3D-Rekonstruktion der Probe nötig.
- Die Lichtscheibe kann auch erzeugt werden, indem ein normaler Laserfokus hoch- und runtergescannt wird.[10] Dieses verfahren ermöglicht es auch selbsterhaltende Laserstrahlen, wie etwa Bessel-Strahlen zu verwenden, die die Eindringtiefe des lightsheets in die Probe deutlich erhöhen, weil der negative Effekt der Streuung an der Probe gemindert wird.[11][12]
- Bei der sog. Oblique Plane Microscopy (OPM)[13] wird das Detektionsobjektiv auch zur Projektion des lightsheets verwendet. Dieses verlässt das Objektiv unter einem Winkel von etwa 60° und zusätzliche Optik im Detektionsstrahlengang des Mikroskops wird eingesetzt, um auch die Fokusebene bzw. Detektionsebene entsprechend zu kippen.
- Eine Fluoreszenzanregung nach dem Zweiphotonen-Prinzip (zwei Photonen doppelter Wellenlänge regen den Fluorophor zusammen an) wurde realisiert. Diese Beleuchtungsmodalität verbessert vor allem die Eindringtiefe in streuende Proben.[14]
- SPIM wurde als Mikroskopietechnik in Verbindung mit Fluoreszenz-Korrelations-Spektroskopie (SPIM-FCS) eingesetzt, um räumlich aufgelöste Mobilitätskarten fluoreszierender Teilchen (z. B. fluoreszierende Mikrosphären, Quanten-Dots oder fluoreszenzmarkierte Proteine) in lebenden biologischen Proben/Organismen zu messen.[15][16][17]
- LSFM wurde auch mit super-resolution-microscopy-Techniken kombiniert, um die Abbe’sche Auflösungsgrenze zu überwinden.[18][19] Auch das stimulated-emission-depletion-Prinzip (STED) wurde für die Lichtscheiben-Beleuchtung implementiert, um die Dicke der Lichtscheibe zu reduzieren und damit die longitudinale Auflösung zu verbessern.[20]
Probenhalterung
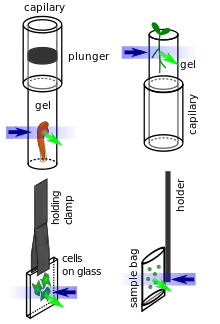
Die Trennung der Beleuchtungs und Detektionsstrahlengängen in den meisten LSFMs und die Tatsache, dass diese meist in einer horizontalen Ebene angeordnet sind, macht spezielle Probenhalterungen notwendig. Die Proben werden oft von oben hängend oder auf einem stehenden Halter montiert (siehe Abbildungen rechts). Für verschiedene Proben wurden verschiedene Halterungen entwickelt:
- Tote (z. B. fixierte) und große Proben können auf einen Halter in der Probenkammer z. B. mit Klebstoff befestigt werden.
- Größere lebende Organismen (Embryos …) können sediert und dann in einen weichen Gelzylinder eingeschlossen werden, der aus einer Glas- oder Plastikkapilare herausgeschoben wird
- Adhärente Zellen lässt man direkt auf kleine Glasplättchen aufwachsen, die dann von oben in die Probenkammer hängen.
- Pflanzen können in klaren Gelen wachsen, wenn die Gele mit einem geeigneten Wachstums- und Nährmedium hergestellt wurden. Die Gele werden typischerweise um die Beobachtungsregion entfernt, damit sie durch Streuung und Absorption die Qualität der Lichtscheibe nicht verschlechtern.[21]
- Flüssige Proben (z. B. für SPIM-FCS) können in kleine Päckchen aus einer dünnen Plastikfolie eingeschweißt werden. Wichtig ist, dass die Plastikfolie denselben Brechungsindex wie das umgebende Medium aufweist, um die Abbildungsleistung des SPIM nicht zu stören.[16][17]
Es wurden auch einige LSFMs entwickelt, die den Anregungs- und Detektionsstrahlengang in einer aufrechten Ebene realisieren. Damit können Proben auch mit mikroskopischen Standardmethoden (z. B. Zellen in einer Petrischale) montiert werden. Auch eine Kombination eines LSFM mit einem darunter liegenden inversen Mikroskop wird möglich.[22][15][23]
Auflösungsvermögen
Die Beobachtung erfolgt bei SPIM über ein Mikroskopobjektiv, welches in die wassergefüllte Probenkammer eintaucht und die Probe direkt abbildet. Damit ist die laterale Auflösung vollständig durch dieses Objektiv gegeben und erreicht maximal etwa eine halbe bis eine Wellenlänge (also z. B. bei grüner Fluoreszenz etwa 250–500 nm).[6] Die axiale Auflösung ist deutlich schlechter (typischerweise um mehr als einen Faktor 4). Sie kann aber etwas verbessert werden, indem das Lichtblatt dünner gemacht wird, sodass nur in einem Teil des Beobachtungsfokus Fluoreszenz angeregt wird. Idealerweise wird so die axiale Auflösung gleich der lateralen.
Im Vergleich mit einem normalen Weitfeldmikroskop ist die axiale Auflösung deutlich besser. Für kleine numerische Aperturen ist die axiale Auflösung sogar besser als bei konfokalen Mikroskopen, bei größeren numerischen Aperturen ist sie noch in einer vergleichbaren Größenordnung.[6] Im Vergleich zur konfokalen Mikroskopie wird das Bild nicht in 3D abgerastert, sondern in Scheiben, von denen jeweils alle Bildpunkte gleichzeitig aufgenommen werden können.
Geschichte
Anfang des 20. Jahrhunderts wurde von R. A. Zsigmondy mit dem Ultramikroskop ein neues Beleuchtungsverfahren in die Dunkelfeldmikroskopie eingeführt. Dabei beleuchtet Sonnenlicht oder eine Weißlichtlampe einen optischen Spalt, der dann mit einer Linse in die Probe abgebildet wird. Kleine Teilchen, die das so gebildete Lichtblatt durchlaufen, können anhand ihres Streulichts unter einem rechten Winkel zur Beleuchtung mit einem Beobachtungsmikroskop beobachtet werden. Dieses Mikroskop erlaubte die Beobachtung von Teilchen kleiner als die optische Auflösung des Beobachtungsmikroskops und führte 1925 zur Vergabe des Nobelpreises an Zsigmondy.[24]
Die erste Anwendung dieses Beleuchtungsprinzips für die Fluoreszenzmikroskopie wurde ab 1993 von Voie et al. unter dem Namen Orthogonal-plane fluorescence optical sectioning (OPFOS) veröffentlicht.[3] Damals zur Abbildung der inneren Struktur der Cochlea mit einer Auflösung von 10 µm lateral und 26 µm longitudinal, allerdings bei einer Probengröße im Millimeterbereich. Zur Formung der Lichtscheibe wurde eine einfache Zylinderlinse verwendet. Eine weitere Entwicklung und Verbesserung des Verfahrens erfolgte dann ab 2004.[4] Danach fand die Technik weite Anwendung und wird bis heute durch neue Varianten angepasst (siehe oben). Seit 2010 sind Ultramikroskope mit Fluoreszenzanregung und niedriger Auflösung[25] und seit 2012 auch SPIM-Mikroskope kommerziell verfügbar.[26] Eine gute Übersicht über die Entwicklung findet sich z. B. in Ref.[27] In den Jahren 2012/2013 wurden erste Open-Source-Projekte zu LSFMs gestartet. Diese veröffentlichen den kompletten Bauplan, incl. der nötigen Software für den Aufbau eines LSFMs.[28][29][30][31]
Anwendung
SPIM wird oft in der Entwicklungsbiologie eingesetzt, wo sie z. B. die Langzeitbeobachtung der embryonalen Entwicklung ermöglicht.[32][4] Sie kann aber auch mit Techniken, wie Fluoreszenzkorrelationsspektroskopie kombiniert werden, um ortsaufgelöste Mobilitätsmessungen fluoreszierender Teilchen (z. B. Beads, Quanten-Dots, fluoreszenzmarkierte Proteine) in (biologischen) Proben zu ermöglichen.[16]
 Brownsche Bewegung von fluoreszierenden Latex-Kügelchen (Durchmesser etwa 20 nm) in Wasser, aufgenommen mit einem SPI-Mikroskop.
Brownsche Bewegung von fluoreszierenden Latex-Kügelchen (Durchmesser etwa 20 nm) in Wasser, aufgenommen mit einem SPI-Mikroskop.
Literatur
- Übersichtsartikel:
- J. Huisken, D.Y.R. Stainier: Selective plane illumination microscopy techniques in developmental biology. In: Development. 136, Nr. 12, 22. Mai 2009, S. 1963–1975. doi:10.1242/dev.022426.
- P. A. Santi: Light Sheet Fluorescence Microscopy: A Review. In: Journal of Histochemistry & Cytochemistry. 59, Nr. 2, 1. Februar 2011, S. 129–138. doi:10.1369/0022155410394857.
Weblinks
- Video eines typischen Experiments aus der Entwicklungsbiologie mit einem SPIM: In dem verlinkten Video wurde die Entwicklung eines Fruchtfliegen-Embryos über ca. 20 Stunden beobachtet. Es zeigt zwei Projektionen des kompletten 3D-Datensatzes.
- OpenSPIM: OpenSPIM ist eine Plattform um die SPIM-Technologie zu bauen, anzupassen und zu verbessern.
- Mikroskopie: Nervenbahnen eines Hühnerembryos (Lichtscheibenmikroskopie)
Einzelnachweise
- Philipp J. Keller, Ernst H. K. Stelzer: Lichtscheiben-Mikroskopie in der molekularen Zellbiophysik In: LABORWELT. 7. Jahrgang, Nr. 5, 2006, S. 18–21 (Online-Version (Memento des Originals vom 20. Januar 2013 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.; PDF; 7,5 MB).
- U. Krzic, S. Günther, L. Hufnagel, D. von Gegerfelt, H. Karlsson, E. Illy, J. Hel: Lichtscheiben-Fluoreszenzmikroskopie (SPIM) und Laser-Anregung in orange zur Abbildung lebender Organismen. In: BioPhotonik. Nr. 1, 2011, S. 42–44 (Online-Version).
- A. H. Voie, D. H. Burns, F. A. Spelman: Orthogonal-plane fluorescence optical sectioning: Three-dimensional imaging of macroscopic biological specimens. In: Journal of Microscopy. 170, Nr. 3, Juni 1993, S. 229–236. doi:10.1111/j.1365-2818.1993.tb03346.x.
- J. Huisken, J. Swoger, F. Del Bene, J. Wittbrodt, E. H. Stelzer: Optical sectioning deep inside live embryos by selective plane illumination microscopy. In: Science. Band 305, Nr. 5686, 2004, S. 1007–1009, doi:10.1126/science.1100035, PMID 15310904.
- Timo Mappes, Norbert Jahr, Andrea Csaki, Nadine Vogler, Juergen Popp, Wolfgang Fritzsche: The Invention of Immersion Ultramicroscopy in 1912-The Birth of Nanotechnology?. In: Angewandte Chemie International Edition. 51, Nr. 45, 5. November 2012, S. 11208–11212. doi:10.1002/anie.201204688.
- K. Greger, J. Swoger, E. H. Stelzer: Basic building units and properties of a fluorescence single plane illumination microscope. In: The Review of scientific instruments. Band 78, Nr. 2, 2007, S. 023705, PMID 17578115.
- Jan Huisken, Didier Y. R. Stainier: Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM). In: Optics Letters. 32, Nr. 17, 2007, S. 2608. doi:10.1364/OL.32.002608.
- Raju Tomer, Khaled Khairy, Fernando Amat, Philipp J Keller: Quantitative high-speed imaging of entire developing embryos with simultaneous multiview light-sheet microscopy. In: Nature Methods. 9, Nr. 7, 3. Juni 2012, S. 755–763. doi:10.1038/nmeth.2062.
- Uros Krzic, Stefan Gunther, Timothy E Saunders, Sebastian J Streichan, Lars Hufnagel: Multiview light-sheet microscope for rapid in toto imaging. In: Nature Methods. 9, Nr. 7, 3. Juni 2012, S. 730–733. doi:10.1038/nmeth.2064.
- P. J. Keller, A. D. Schmidt, J. Wittbrodt, E. H.K. Stelzer: Reconstruction of Zebrafish Early Embryonic Development by Scanned Light Sheet Microscopy. In: Science. 322, Nr. 5904, 14. November 2008, S. 1065–1069. doi:10.1126/science.1162493.
- F. O. Fahrbach, A. Rohrbach: A line scanned light-sheet microscope with phase shaped self-reconstructing beams. In: Optics express. Band 18, Nummer 23, November 2010, S. 24229–24244, PMID 21164769.
- T. A. Planchon, L. Gao, D. E. Milkie, M. W. Davidson, J. A. Galbraith, C. G. Galbraith, E. Betzig: Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. In: Nature methods. Band 8, Nummer 5, Mai 2011, S. 417–423, doi:10.1038/nmeth.1586. PMID 21378978.
- C. Dunsby: Optically sectioned imaging by oblique plane microscopy. In: Optics Express. 16, Nr. 25, 2008, S. 20306. doi:10.1364/OE.16.020306.
- Zeno Lavagnino, Francesca Cella Zanacchi, Emiliano Ronzitti, Alberto Diaspro: Two-photon excitation selective plane illumination microscopy (2PE-SPIM) of highly scattering samples: characterization and application. In: Optics Express. 21, Nr. 5, 2013, S. 5998. doi:10.1364/OE.21.005998.
- J. Capoulade, M. Wachsmuth, L. Hufnagel, M. Knop: Quantitative fluorescence imaging of protein diffusion and interaction in living cells. In: Nature Biotechnology. Band 29, Nummer 9, September 2011, S. 835–839, doi:10.1038/nbt.1928. PMID 21822256.
- T. Wohland, X. Shi, J. Sankaran, E. H. Stelzer: Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments. In: Optics express. Band 18, Nr. 10, 2010, S. 10627–10641, PMID 20588915.
- Jan W Krieger, Anand P Singh, Nirmalya Bag, Christoph S Garbe, Timothy E Saunders, Jörg Langowski, Thorsten Wohland: Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. In: Nature Protocols. Band 10, Nr. 12, 5. November 2015, S. 1948, doi:10.1038/nprot.2015.100 (uni-heidelberg.de [PDF]).
- Francesca Cella Zanacchi, Zeno Lavagnino, Michela Perrone Donnorso, Alessio Del Bue, Laura Furia, Mario Faretta, Alberto Diaspro: Live-cell 3D super-resolution imaging in thick biological samples. In: Nature Methods. 8, Nr. 12, 9. Oktober 2011, S. 1047–1049. doi:10.1038/nmeth.1744.
- Jerome Mertz, Jinhyun Kim: Scanning light-sheet microscopy in the whole mouse brain with HiLo background rejection. In: Journal of Biomedical Optics. 15, Nr. 1, 2010, S. 016027. doi:10.1117/1.3324890.
- M. Friedrich, Q. Gan, V. Ermolayev, G. S. Harms: STED-SPIM: Stimulated emission depletion improves sheet illumination microscopy resolution. In: Biophysical Journal. Band 100, Nummer 8, April 2011, S. L43–L45, doi:10.1016/j.bpj.2010.12.3748. PMID 21504720. PMC 3077687 (freier Volltext).
- Alexis Maizel, Daniel von Wangenheim, Fern n Federici, Jim Haseloff, Ernst H.K. Stelzer: High-resolution live imaging of plant growth in near physiological bright conditions using light sheet fluorescence microscopy. In: The Plant Journal. 68, Nr. 2, Oktober 2011, S. 377–385. doi:10.1111/j.1365-313X.2011.04692.x.
- Terrence F. Holekamp, Diwakar Turaga, Timothy E. Holy: Fast Three-Dimensional Fluorescence Imaging of Activity in Neural Populations by Objective-Coupled Planar Illumination Microscopy. In: Neuron. 57, Nr. 5, 13. März 2008, S. 661–672. doi:10.1016/j.neuron.2008.01.011.
- Y. Wu, A. Ghitani, R. Christensen, A. Santella, Z. Du, G. Rondeau, Z. Bao, D. Colon-Ramos, H. Shroff: Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. In: Proceedings of the National Academy of Sciences. 108, Nr. 43, 25. Oktober 2011, S. 17708–17713. doi:10.1073/pnas.1108494108.
- Nobelpreis-Vorlesung von R. A. Zsigmondy (englisch): Properties of colloids (PDF; 108 kB), mit einer Abbildung und kurzen Erklärung zum Ultramikroskop
- Presseveröffentlichung von LaVision Biotech (Memento vom 24. Dezember 2013 im Internet Archive) (abgerufen am 4. November 2012)
- Carl Zeiss-Presseveröffentlichung zum Lichtblattmikroskopsystem Lightsheet Z.1 (abgerufen am 4. November 2012)
- P. A. Santi: Light Sheet Fluorescence Microscopy: A Review. In: Journal of Histochemistry & Cytochemistry. 59, Nr. 2, 1. Februar 2011, S. 129–138. doi:10.1369/0022155410394857.
- OpenSPIM project webpage (abgerufen am 8. Juni 2013)
- Peter G Pitrone, Johannes Schindelin, Luke Stuyvenberg, Stephan Preibisch, Michael Weber, Kevin W Eliceiri, Jan Huisken, Pavel Tomancak: OpenSPIM: an open-access light-sheet microscopy platform. In: Nature Methods. 9. Juni 2013. doi:10.1038/nmeth.2507.
- The OpenSPIN project webpage (abgerufen am 8. Juni 2013)
- Emilio J Gualda, Tiago Vale, Pedro Almada, Jos A Feij, Gabriel G Martins, Nuno Moreno: OpenSpinMicroscopy: an open-source integrated microscopy platform. In: Nature Methods. 9. Juni 2013. doi:10.1038/nmeth.2508.
- P. J. Verveer, J. Swoger, F. Pampaloni, K. Greger, M. Marcello, E. H. Stelzer: High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. In: Nature methods. Band 4, Nr. 4, 2007, S. 311–313, doi:10.1038/nmeth1017, PMID 17339847.
- Corinne Lorenzo, Céline Frongia, Raphael Jorand, Jérome Fehrenbach, Pierre Weiss, Amina Maandhui, Guillaume Gay, Bernard Ducommun, Valérie Lobjois: Live cell division dynamics monitoring in 3D large spheroid tumor models using light sheet microscopy. In: Cell Division. 6, Nr. 1, 2011, S. 22. doi:10.1186/1747-1028-6-22.
- Daisuke Takao, Atsushi Taniguchi, Takaaki Takeda, Seiji Sonobe, Shigenori Nonaka, Alexandre J. Kabla: High-Speed Imaging of Amoeboid Movements Using Light-Sheet Microscopy. In: PLoS ONE. Band 7, Nr. 12, 5. Dezember 2012, S. e50846, doi:10.1371/journal.pone.0050846.