Multiphotonenmikroskop
Ein Multiphotonenmikroskop (englisch Multi-Photon Laser Scanning Microscope, MPLSM, auch multi-photon microscopy, MPM[1][2]) ist ein spezielles Lichtmikroskop aus der Gruppe der Laser-Scanning-Mikroskope.
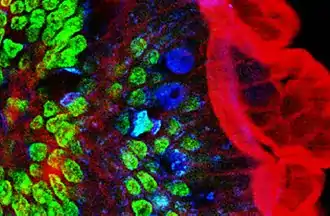
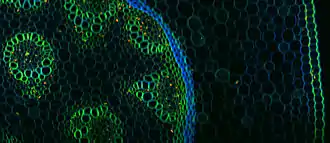
Bilder werden erzeugt, indem eines von zwei unterschiedlichen physikalischen Phänomenen ausgenutzt wird:
- Multiphotonen-Fluoreszenz (meist Zwei-Photonen-Fluoreszenz) oder
- Higher Harmonic Generation (Verdopplung (Second Harmonic Generation, SHG) oder Verdreifachung (Third Harmonic Generation, THG) der Schwingungsfrequenz des eingestrahlten Lichtes).
Mit Hilfe eines starken, fokussierten Laserstrahls werden dabei nichtlineare optische Effekte erzeugt, die auf dem Zusammenspiel mehrerer gleichzeitig in einem Molekül eintreffender Photonen (Lichtteilchen) beruhen. Die Stärke des erzeugten Signals steigt daher nicht linear mit der Zahl der eingestrahlten Photonen, sondern mit dem Quadrat (bei Zwei-Photonen-Effekten) oder der dritten Potenz (bei Drei-Photonen-Effekten).
Die Arbeitsweise eines Multiphotonenmikroskops ähnelt der eines konfokalen Laser-Scanning-Mikroskops. Während jedoch die konfokale Laser-Scanning-Mikroskopie eine Eindringtiefe je nach Präparat von 50–80 µm hat, können mit der Multi-Photonen-Mikroskopie tiefere Bereiche, z. B. von 200 µm, in sehr günstigen Fällen sogar bis zu 1000 µm (=1 mm) erreicht werden.[3] Dadurch sind Aufnahmen von lebenden Geweben möglich, die anderweitig für die Bildgebung unerreichbar sind.
Die erreichbare Auflösung von Multiphotonenmikroskopen ist im Artikel Auflösung (Mikroskopie) im Abschnitt Multi-Photonen-Anregung beschrieben.
Multi-Photonen-Fluoreszenzmikroskopie

Die am weitesten verbreitete Multiphotonentechnik ist die Zwei-Photonen-Fluoreszenzmikroskopie, manchmal auch nur Zweiphotonenmikroskopie genannt. Bei der herkömmlichen Fluoreszenzmikroskopie wird in einem fluoreszierenden Molekül ein Elektron durch die Absorption jeweils eines Photons angeregt, also in einen höheren Energiezustand versetzt. Bei der Zwei-Photonen-Fluoreszenzmikroskopie wird die Anregung des Elektrons durch die gleichzeitige Absorption zweier Photonen hervorgerufen (Zwei-Photonen-Absorption). Auch eine Anregung mit drei oder mehr gleichzeitig eintreffenden Photonen ist möglich.
Prinzip
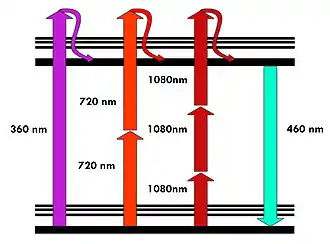
Fluoreszenz entsteht, wenn Farbstoffe ankommende Photonen absorbieren und in der Folge ein anderes Photon wieder abgeben. Durch das ankommende, „anregende“ Photon wird ein Elektron auf ein höheres Energieniveau gehoben, die Energie also derart zwischengespeichert. Bei normaler Fluoreszenzmikroskopie geschieht diese Anregung durch genau ein Photon. Das Elektron bleibt für einige Nanosekunden auf dem höheren Energieniveau, bevor es wieder zurückfällt und dabei ein neues, längerwelliges, energieärmeres Photon aussendet. Wenn etwa mit blauem Licht angeregt wird, entsteht meist grüne Fluoreszenz, beispielsweise bei Fluorescein.
Dieses einzelne Anregungsphoton kann durch zwei oder mehr Photonen ersetzt werden, wenn diese in der Summe die gleiche Energie haben. So kann dunkelrotes oder infrarotes Licht eingesetzt werden, um grüne Fluoreszenz zu erzeugen. Außerdem müssen beide Photonen gleichzeitig (innerhalb einer Attosekunde = 10−18 s) eintreffen, da kein stabiles Zwischenenergieniveau existiert.
Bei der normalen Fluoreszenzmikroskopie hat das anregende Photon eine kürzere Wellenlänge, d. h. eine höhere Frequenz und somit mehr Energie als das abgestrahlte Photon. Die Wellenlängen-Differenz der beiden Photonen wird als Stokes-Shift bezeichnet. Im Gegensatz dazu wird bei der Multi-Photonen-Anregung mit Photonen angeregt, die eine deutlich größere Wellenlänge, niedrigere Frequenz und somit pro Photon weniger Energie haben, als die ausgesandten Photonen. Dies ist nur möglich, weil hier zwei oder mehr anregende Photonen zur Erzeugung nur eines ausgesandten Photons führen. Bei der Zwei-Photonen-Anregung beträgt die Anregungswellenlänge in etwa das Doppelte der normalerweise verwendeten Anregungswellenlänge, bei Drei-Photonen-Anregung ein Dreifaches.[4][5]
Technische Umsetzung
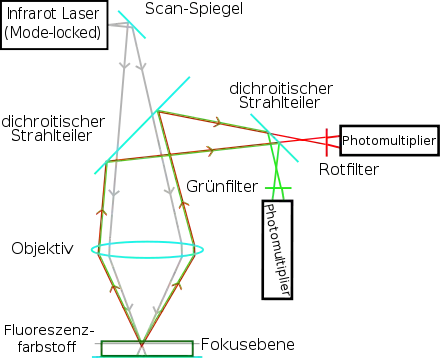
Um ein gleichzeitiges Eintreffen zweier oder mehr Photonen bei den anregbaren Elektronen im Fokuspunkt zu erreichen, sind sehr hohe Photonendichten erforderlich. Diese werden nur erzielt, wenn ein gepulster Laser mit Modenkopplung eingesetzt wird. Das Besondere an diesem Lasertyp ist, dass sehr kurze (z. B. 0,14 ps = 0,14·10−12 s), intensive Laserpulse ausgesandt werden, die z. B. 80 Millionen Mal pro Sekunde wiederholt werden. Die Pausen zwischen zwei Pulsen sind im gegebenen Beispiel also 12,5 ns (= 12.500 ps) lang, so dass die gesamte im Laser erzeugte Energie in einem Bruchteil der Zeit abgegeben werden kann.[6]
Die für die Anregung in der Regel eingesetzten Titan:Saphir-Laser sind kostspielig (ca. 150.000 Euro) und stellen daher eine große Hürde für einen verbreiteten Einsatz dar. Ti:Sa-Laser können auf Wellenlängen von etwa 700 nm bis etwa 1050 nm eingestellt werden. Größere Wellenlängen können durch den Einsatz eines „optisch parametrischen Oszillators“ (OPO) erzeugt werden. Dieser wird mit dem Ti:Sa-Laser „gepumpt“ und kann dann Wellenlängen bis über 1300 nm erzeugen. Damit können auch rote und dunkelrote Fluoreszenzfarbstoffe im Zwei-Photonen-Modus angeregt werden.
Wie bei einem konfokalen Laser-Scanning-Mikroskop wird der Laserstrahl durch das Objektiv des Mikroskops auf einen Punkt des Präparats fokussiert. Durch im Strahlengang befindliche bewegliche Spiegel (Scanspiegel; engl. to scan = abrastern) wird der Laserstrahl in seiner Lage so verändert, dass der Fokuspunkt sich durch das Präparat bewegt, dieses also abrastert. Die dadurch entstehende Fluoreszenz wird vom Objektiv aufgefangen, über dichroitische Strahlteiler spektral aufgetrennt und zum Detektor geleitet (vorwiegend Photomultiplier, aber auch Avalanche-Photodioden[7][8]). Die Detektoren messen die Helligkeit jedes Bildpunktes also nacheinander. Zu keinem Zeitpunkt entsteht im Mikroskop ein vollständiges Bild des Präparats, dies wird erst im Steuerungscomputer zusammengesetzt.[4][5]
Auf Grund der Komplexität werden Komplettgeräte derzeit nur von wenigen Herstellern angeboten. Da die Scanning- und Detektortechnik eines Multiphotonenmikroskops der eines konfokalen Laser-Scanning-Mikroskops sehr ähnlich ist, gibt es einige Firmen, die entsprechende Erweiterungen anbieten, sowie Arbeitsgruppen, die solche Mikroskopie in Eigenregie mit einem geeigneten Anregungslaser und den entsprechenden Filtern zum Multiphotonenmikroskop aufrüsten.
Vorteile
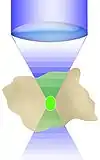
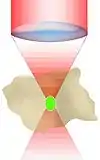
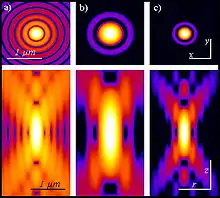
Wie oben dargestellt, erfordert die Erzeugung des Zwei-Photonen-Effekts eine sehr hohe Photonendichte, die nur ein gepulster Laser erzielen kann. Aber auch in diesem Fall kommt es nur im Fokuspunkt zu einer genügend hohen Photonendichte, um eine Fluoreszenzanregung zu erzeugen, nicht aber darüber und darunter (siehe Abbildung): Außerhalb der Fokusebene verteilt sich die gleiche Menge Anregungsphotonen auf einen stark zunehmenden Durchmesser des Strahlkegels. Zwei-Photonen-Anregung hängt aber vom Quadrat der Lichtintensität ab, so dass die Lichtintensitäten außerhalb der Fokusebene, im Gegensatz zu anderen Fluoreszenzmikroskopen, für die Erzeugung von Fluoreszenz nicht mehr ausreicht.[5][9]
Daraus ergeben sich praktische Vorteile:
- Ein Ausbleichen von Fluoreszenzfarbstoffen und die Erzeugung von Phototoxizität ist auf eine extrem kleine Umgebung des Fokuspunktes beschränkt. Ebenen darüber und darunter sind nicht betroffen.
- Die gesamte vom Objektiv aufgefangene Fluoreszenz kann für das zu erstellende Bild verwendet werden. Im Gegensatz zum konfokalen Laser-Scanning-Mikroskop ist also keine Lochblende (pinhole) nötig, um Licht aus anderen Ebenen auszufiltern. Daher ist es, wiederum im Vergleich zum konfokalen Laser-Scanning-Mikroskop, auch nicht nötig, die Fluoreszenz über die Scanspiegel aufzufangen. Stattdessen kann eine „non-descanned detection“ durchgeführt werden. Die Detektion kann dadurch räumlich dichter am Präparat erfolgen, was wiederum das Auffangen eines Teils der im Präparat gestreuten Fluoreszenz erlaubt.
- Ein davon unabhängiger Vorteil ist die höhere Eindringtiefe durch die geringere Streuung von längerwelligem Licht. Der Streuquerschnitt σ hängt sehr stark von der Frequenz ν ab und steigt proportional zu ν4. Kurzwelliges violettes Licht (400 nm) hat eine doppelt so hohe Frequenz wie langwelliges rotes Licht (800 nm) und wird daher 16-mal stärker gestreut (siehe Das Blau beziehungsweise das Rot des Himmels). Wellenlängenabhängige Streuung geschieht auch in biologischen Geweben: Wenn mit einer starken Taschenlampe durch eine Hand geleuchtet wird, dringt fast nur der rote Lichtanteil durch. Da bei der Zwei-Photonen-Mikroskopie infrarotes oder dunkelrotes Licht für die Fluoreszenzanregung eingesetzt wird, können entsprechend tiefere Regionen erreicht werden.[4]
Higher Harmonic Generation
Neben Fluoreszenz spielen in der Multiphotonenmikroskopie Second und Third Harmonic Generation (SHG bzw. THG; wörtlich: Erzeugung der zweiten (bzw. dritten) Harmonischen; im Deutschen auch: Frequenzverdopplung bzw. Frequenzverdreifachung) eine Rolle. Sie werden als Higher Harmonic Generation (HHG) zusammengefasst.
Diese Erzeugung von Licht mit niedrigerer Wellenlänge ist physikalisch nicht verwandt mit Multiphotonen-Fluoreszenzanregung. HHG tritt aber unter gleichartigen Beleuchtungsbedingungen auf wie Multiphotonenfluoreszenz, nämlich (nur) bei sehr starkem Anregungslicht. Auch HHG-Signale entstehen daher nur im Fokuspunkt eines gepulsten Lasers, aber nicht darüber oder darunter. Die oben dargestellten Abschnitte ‚Technische Umsetzung‘ und ‚Vorteile‘ gelten entsprechend. Die erforderliche technische Ausstattung ist weitgehend gleich, so dass beispielsweise ein Gerät, das für Zwei-Photonen-Fluoreszenzmikroskopie gebaut wurde, meistens auch Second Harmonic Generation ermöglicht.
Grundlagen
Licht ist eine elektromagnetische Strahlung, entsprechend hat es ein elektrisches Feld. Dieses Feld tritt in Wechselwirkungen mit der durchstrahlten Materie. Wenn im Multiphotonenmikroskop der gepulste Laser auf ein Präparat fokussiert wird, führen diese Wechselwirkungen zur Entstehung von „Harmonischen“. Die Wellenlänge der „Second Harmonic“ ist exakt die Hälfte des eingestrahlten Lichtes, die der „Third Harmonic“ bei THG exakt ein Drittel.[10][11]
Im Gegensatz zur Fluoreszenz bleibt bei HHG keine Energie im Präparat zurück, auch ein Ausbleichen des Signals kommt nicht vor. Phototoxische Effekte können jedoch unabhängig vom HHG-Effekt durch die gleichzeitige Anregung von Autofluoreszenz oder durch Absorption entstehen. Auch kann eine zu hohe Intensität des Anregungslichts direkt zur Zerstörung von Präparaten führen.[10][11]
Second Harmonic Generation


Die Erzeugung eines SHG-Signals, also Frequenzverdopplung, ist nur möglich, wenn sich die elektrischen Eigenschaften des bestrahlten Moleküls in allen Raumrichtungen unterscheiden, wenn es also asymmetrisch, genauer nicht-centrosymmetrisch, ist.
Das SHG-Signal breitet sich hauptsächlich in der „Vorwärts“-Richtung aus, wie der eintreffende Lichtstrahl auch: Die Einzelphasen der nach vorn gerichteten Photonen (das SHG-Signal) sind meist phasengleich (kohärent), so dass sich die Wellen, die von verschiedenen Molekülen erzeugt werden, verstärken. In anderen Richtungen löschen sich die Wellen teilweise gegenseitig aus (destruktive Interferenz). Die Anteile des nach Vorwärts und des nach Rückwärts gerichteten Signals hängen auch von der Struktur der bestrahlten Moleküle ab. Die Stärke des entstehenden Signals ist ferner abhängig von der Polarisationsrichtung des eintreffenden Laserlichts. Bei Objekten mit Längsstruktur (z. B. Muskelfasern) ergibt sich dadurch eine Abhängigkeit der Signalstärke von der Orientierung der Polarisationsebene des Lasers zum Präparat.[10][11]
Aus dem Entstehungsmechanismus ergibt sich, dass SHG an manchen periodischen Strukturen besonders effizient erzeugt wird, z. B. an Harnstoffkristallen. In biologischen Geweben entsteht es etwa an Kollagenfasern und am Myosin in glatter Muskulatur. SHG erleichtert dadurch die Orientierung im Präparat, auch wenn hauptsächlich Fluoreszenz beobachtet werden soll.[10][11]
Da das kurzwelligste Licht, das an einem Mikroskop aufgezeichnet werden kann, in der Regel im blauen Bereich liegt, wird für die Erzeugung von SHG-Signalen eine Wellenlänge von über 800 nm eingesetzt.
Third Harmonic Generation
Wenn die Anregungswellenlänge über 1200 nm liegt, lässt sich auch Third Harmonic Generation (THG; Frequenzverdreifachung) beobachten beziehungsweise mit Filtern für sichtbares Licht auffangen. Im Gegensatz zu SHG ist THG nicht auf das Vorhandensein nicht-centrosymmetrischer Strukturen angewiesen. Bei genügend hoher Intensität des eingestrahlten Lichtes kann im Prinzip in jedem Stoff THG hervorgerufen werden, die Stärke des Signals hängt jedoch vom Material ab (siehe Frequenzverdopplung). Kontrastreiche THG-Bilder entstehen, wenn optisch unterschiedlich dichte Strukturen nebeneinander liegen, beispielsweise Zellen und Blutplasma.[9][10]
Geschichte und Anwendungen
1931–1990
Das physikalische Prinzip der Fluoreszenz-Anregung eines Moleküls durch mehrere Photonen wurde zuerst 1931 von Maria Goeppert-Mayer vorhergesagt.[12] Die erste experimentelle Beobachtung von Zwei-Photonen-Fluoreszenzanregung erfolgte 1961, bald nach Entwicklung der ersten Laser.[5] Mikroskopie mit Zwei-Photonen-Fluoreszenz-Anregung gelang 1990 das erste Mal.[13]
Der SHG-Effekt wurde direkt nach der Entwicklung des Lasers 1960 beobachtet. Mikroskopisch wurde er 1974 erstmals eingesetzt, zunächst in einem konventionellen Lichtmikroskop ohne Scanning-Technik: Robert Hellwarth und Paul Christensen (University of Southern California, Los Angeles) um die Struktur von Zinkselenid-Polykristallen zu untersuchen.[14] 1978 wurde SHG von J. N. Gannaway und C. J. R. Sheppard erstmals mit einem Scanning-Mikroskop erzeugt. Sie waren somit die ersten, die das erzeugte Signal auf die Fokusebene beschränken konnten. Auch sie untersuchten Kristalle.[15] Die dabei eingesetzten kontinuierlich strahlenden Laser setzen aber im Präparat so viel Energie frei, dass biologische Präparate zerstört werden. Erst mit der Einführung gepulster Laser wurde dieses Problem gelöst, weil nur damit der mittlere Energieeintrag ausreichend gering ist.
Die vermutlich erste Anwendung von SHG an einem biologischen Präparat gelang 1980 der Arbeitsgruppe um Isaac Freund an der Bar-Ilan-Universität in Ramat-Gan, Israel, indem sie Kollagen in Sehnen von Ratten untersuchten. Dabei wurde ein nd:YAG-Laser mit einer festen Wellenlänge von 1064 nm eingesetzt und das Signal in Vorwärts-Richtung aufgefangen. Zunächst konnte zwar die Intensität in Abhängigkeit vom Einfallswinkel des Laserstrahls gemessen werden, ein Bild konnte jedoch zunächst noch nicht erstellt werden.[16] Dies gelang der gleichen Arbeitsgruppe jedoch am gleichen Objekt 1986.[17]
Seit 1990
Nachdem Denk et al. 1990 das erste Mal Zweiphotonen-Fluoreszenzmikroskopie demonstriert hatten,[13] dauerte es nur vier weitere Jahre, bis es gelang, sie an lebenden Tieren durchzuführen (Intravitalmikroskopie), in diesem Fall um den Blutfluss und das Verhalten von weißen Blutkörperchen in der Niere zu untersuchen.[18]
Die Vorteile eines Multiphotonenmikroskops, speziell die hohe Eindringtiefe, kommen besonders in Geweben zum Tragen, bei denen strukturelle Unterschiede zwischen den oberen und den tieferen Gewebeschichten vorliegen: Die tieferen Gewebeschichten sind für andere Arten der Mikroskopie nicht bzw. nur in fixierten, geschnittenen Präparaten zugänglich. Dortige Vorgänge können daher in lebenden Organen nicht anders beobachtet werden. Beispiele sind Lebenduntersuchungen in verschiedenen Hirnschichten,[19] die Beobachtung von verschiedenen Zellen des Immunsystems in Lymphknoten,[1][2] Untersuchungen, wie Tumorzellen in benachbarte Gewebe eindringen können[20] und Untersuchungen an Muskelzellen im intakten Herzen.[21] In den genannten Beispielen wurde jeweils Zwei-Photonen-Fluoreszenz und teilweise zusätzlich SHG eingesetzt.
Während die Beobachtung von Fluoreszenz in flachen Präparaten (einzelne Zellen, Gewebeschnitte) auch gut in normalen Fluoreszenzmikroskopen oder in konfokalen Laser-Scanning-Mikroskopen stattfinden kann, ist HHG ausschließlich mit einem Multiphotonenmikroskop möglich. Neben den bereits erwähnten Kollagenfasern und Muskelmyosin führen auch Stärke und in schwächerem Maße Cellulose zu SHG.[9]
Daneben ist es möglich, spezifische Farbstoffe einzusetzen, die SHG hervorrufen und beispielsweise Biomembranen anfärben.[9] 1996 wurde mit einem solchen Farbstoff, der empfindlich auf das Membranpotential reagiert, das erste Mal SHG an lebenden Zellen veröffentlicht.[22] Bei anderen SHG-Membranfarbstoffen geht das SHG-Signal verloren, wenn zwei derartig markierte Membranen zusammenkommen, da plötzlich eine Centrosymmetrie auftritt. Dieser Vorgang kann daher sehr empfindlich festgestellt werden.[23]
SHG-Mikroskopie litt lange Zeit an sehr langen Aufnahmezeiten von Minuten bis Stunden pro Bild. Dies änderte sich erst 1999, als es gelang, SHG auf einem Laser-Scanning-Mikroskop zu erzeugen, das mit einem Ti:Sa-Laser ausgestattet war.[24][25]
THG wurde bisher in nur wenigen veröffentlichten biomedizinischen Studien verwendet. Ein Grund hierfür ist, dass bei herkömmliche Titan:Saphir-Laser, die in der Regel für Multiphotonenmikroskope eingesetzt werden, die maximale verfügbare Wellenlänge (unter 1100 nm) nicht ausreicht, um THG im sichtbaren Bereich zu erzeugen. UV-Licht kann jedoch mit der üblichen Geräteausstattung nicht aufgenommen werden, so dass für THG andere Anregungslaser eingesetzt werden. Bisherige Anwendungen waren beispielsweise die Beobachtung von Fetttröpfchen oder die Beobachtung von Hydroxylapatit-Kristallen in Zahnschmelz.[9] Bei einer Anregungswellenlänge oberhalb 1200 nm ist die Beobachtung von THG-Signalen im blauen und SHG-Signalen im roten Teil des Spektrums möglich. Dies wurde beispielsweise genutzt, um intakte Mausembryonen dreidimensional darzustellen.[26]
Weblinks
- Direkter Blick ins Gehirn: Zwei-Photonen-Mikroskopie revolutioniert die Gehirnforschung. In: Scinexx – Das Wissensmagazin.
- Multiphoton Fluorescence Microscopy. In: Microscopy Primer. Florida State University (englisch).
- Multiple-photon excitation fluorescence microscopy. University of Wisconsin (englisch).
- Fundamentals and Applications in Multiphoton Excitation Microscopy. Nikon MicroscopyU (englisch).
- Mehr Licht! Das Mikroskop der Zukunft (deutsch).
Einzelnachweise
- C. Sumen, T. R. Mempel, I. B. Mazo, U. H. von Andrian: Intravital microscopy: visualizing immunity in context. In: Immunity. Band 21, Nr. 3, September 2004, S. 315–329, doi:10.1016/j.immuni.2004.08.006, PMID 15357943.
- P. Friedl, A. T. den Boer, M. Gunzer: Tuning immune responses: diversity and adaptation of the immunological synapse. In: Nat. Rev. Immunol. Band 5, Nr. 7, Juni 2005, S. 532–545, doi:10.1038/nri1647, PMID 15999094.
- Patrick Theer, Mazahir T. Hasan, Winfried Denk: Two-photon imaging to a depth of 1000 µm in living brains by use of a Ti:Al2O3 regenerative amplifier. In: Optics Letters. Band 28, Nr. 12, 2003, S. 1022–1024 (Abstract).
- Alberto Diaspro, Paolo Bianchini, Giuseppe Vicidomini, Mario Faretta, Paola Ramoino, Cesare Usai: Multi-photon excitation microscopy. In: BioMedical Engineering OnLine. Band 5, Nr. 1, 2006, S. 36, doi:10.1186/1475-925X-5-36, PMID 16756664, PMC 1550243 (freier Volltext).
- W. Denk, D. W. Piston, W. W. Webb: Multi-Photon Molecular Excitation in Laser-Scanning Microscopy. In: James B. Pawley (Hrsg.): Handbook of biological Confocal Microscopy. 3. Auflage. Springer, New York NY 2006, ISBN 0-387-25921-X.
- Datenblatt des Chameleon Ultra II (PDF; 363 kB) der Firma Coherent.
- Barry R. Masters: Confocal Microscopy And Multiphoton Excitation Microscopy: The Genesis of Live Cell Imaging. SPIE Press, Bellingham, Washington 2006, ISBN 978-0-8194-6118-6, Chapter 12: Theory and Instrumentation of Multiphoton Excitation Microscopy, S. 169–177.
- Peter T. C. So, Chen Y. Dong, Barry R. Masters, Keith M. Berland: Two-photon excitation fluorescence microscopy. (PDF; 4,7 MB). In: Annu. Rev. Biomed. Eng. Nr. 2, 2000, S. 399–429.
- Peter Friedl, Katarina Wolf, Gregory Harms, Ulrich H. von Andrian: Biological second and third harmonic generation microscopy. In: Curr Protoc Cell Biol. Chapter 4, März 2007, Unit 4.15, doi:10.1002/0471143030.cb0415s34, PMID 18228516.
- Guy Cox, Eleanor Kable: Second-Harmonic Imaging of Collagen. In: Cell Imaging Techniques. Humana Press, Totowa NJ 2006, ISBN 1-58829-157-X, S. 15–35, doi:10.1007/978-1-59259-993-6_2.
- Guy Cox: Optical Imaging Techniques in Cell Biologiy. CRC Press, Taylor & Francis Group, Boca Raton, FL 2007, ISBN 978-0-8493-3919-6, Chapter 8: Nonlinear Microscopy, S. 101–114.
- M. Göppert-Mayer: Über Elementarakte mit zwei Quantensprüngen. In: Ann Phys. Band 9, 1931, S. 273–295, doi:10.1002/andp.19314010303.
- W. Denk, J. H. Strickler, W. W. Webb: Two-photon laser scanning fluorescence microscopy. In: Science. Band 248, Nr. 4951, April 1990, S. 73–76, doi:10.1126/science.2321027, PMID 2321027.
- R. Hellwarth, P. Christensen: Nonlinear optical microscopic examination of structure in polycrystalline ZnSe. In: Optics Communications. Band 12, Nr. 3, November 1974, S. 318–322, doi:10.1016/0030-4018(74)90024-8.
- J. N. Gannaway, C. J. R. Sheppard: Second-harmonic imaging in the scanning optical microscope. In: Optical and Quantum Electronics. Band 10, Nr. 5, September 1978, S. 435–439, doi:10.1007/BF00620308.
- Shmuel Roth, Isaac Freund: Optical second-harmonic scattering in rat-tail tendon. In: Biopolymers. Band 20, Nr. 6, 1981, S. 1271–1290, doi:10.1002/bip.1981.360200613.
- I. Freund, M. Deutsch, A. Sprecher: Connective tissue polarity. Optical second-harmonic microscopy, crossed-beam summation, and small-angle scattering in rat-tail tendon. In: Biophys. J. Band 50, Nr. 4, Oktober 1986, S. 693–712, doi:10.1016/S0006-3495(86)83510-X, PMID 3779007, PMC 1329848 (freier Volltext).
- Kenneth W. Dunn, Ruben M. Sandoval, Katherine J. Kelly, Pierre C. Dagher, George A. Tanner, Simon J. Atkinson, Robert L. Bacallao, Bruce A. Molitoris: Functional studies of the kidney of living animals using multicolor two-photon microscopy. In: Am. J. Physiol., Cell Physiol. Band 283, Nr. 3, 2002, S. C905–C916, doi:10.1152/ajpcell.00159.2002, PMID 12176747 (archiviert am 29. Oktober 2013 [PDF]).
- Karel Svoboda, Ryohei Yasuda: Principles of two-photon excitation microscopy and its applications to neuroscience. In: Neuron. Band 50, Nr. 6, Juni 2006, S. 823–839, doi:10.1016/j.neuron.2006.05.019, PMID 16772166.
- Stephanie Alexander, Gudrun E. Koehl, Markus Hirschberg, Edward K. Geissler, Peter Friedl: Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. In: Histochem. Cell Biol. Band 130, Nr. 6, Dezember 2008, S. 1147–1154, doi:10.1007/s00418-008-0529-1, PMID 18987875.
- John A. Scherschel, Michael Rubar: Cardiovascular imaging using two-photon microscopy. In: Microsc. Microanal. Band 14, Nr. 6, Dezember 2008, S. 492–506, doi:10.1017/S1431927608080835, PMID 18986603.
- I. Ben-Oren, G. Peleg, A. Lewis, B. Minke, L. Loew: Infrared nonlinear optical measurements of membrane potential in photoreceptor cells. In: Biophys. J. Band 71, Nr. 3, September 1996, S. 1616–1620, doi:10.1016/S0006-3495(96)79365-7, PMID 8874036, PMC 1233629 (freier Volltext).
- L. Moreaux, O. Sandre, J. Mertz: Membrane imaging by second-harmonic generation microscopy. In: J. Opt. Soc. Am. B. Band 17, 2000, S. 1685–1694, doi:10.1364/JOSAB.17.001685.
- Paul J. Campagnola, Leslie M. Loew: Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. In: Nat. Biotechnol. Band 21, Nr. 11, November 2003, S. 1356–1360, doi:10.1038/nbt894, PMID 14595363.
- Paul J. Campagnola, Mei-de Wei, Aaron Lewis, Leslie M. Loew: High-resolution nonlinear optical imaging of live cells by second harmonic generation. In: Biophys. J. Band 77, Nr. 6, Dezember 1999, S. 3341–3349, doi:10.1016/S0006-3495(99)77165-1, PMID 10585956, PMC 1300605 (freier Volltext).
- Cho-Shuen Hsieh, Shee-Uan Chen, Yen-Wei Lee, Yu-Shih Yang, and Chi-Kuang Sun: Higher harmonic generation microscopy of in vitro cultured mammal oocytes and embryos. In: Opt Express. Band 16, Nr. 15, Juli 2008, S. 11574–11588, PMID 18648479.