STED-Mikroskop
Ein STED-Mikroskop (STED = Stimulated Emission Depletion) ist eine besondere Form des Lichtmikroskops, dessen Auflösung nicht beugungsbegrenzt ist. Es kann daher noch Strukturen unterscheiden, die deutlich enger beieinander liegen, als es das von Ernst Abbe formulierte Limit der normalen lichtmikroskopischen Auflösungsgrenze angibt. STED ist eine von mehreren Techniken, die eine solche erhöhte Auflösung erlauben (siehe RESOLFT-Mikroskopie).
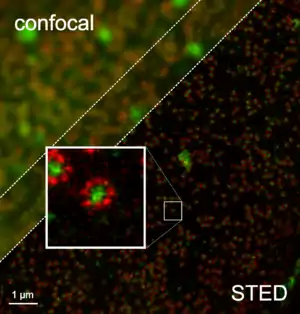
Wie alle derartigen Verfahren ist auch STED eine Spielart der Fluoreszenzmikroskopie, es setzt also die Verwendung von Fluoreszenz-Farbstoffen voraus. Diese sogenannten Fluorochrome lassen sich durch Licht bestimmter Wellenlängen anregen und strahlen anschließend spontan, innerhalb einiger Nanosekunden, Licht über einen Bereich längerer, energieärmerer Wellenlängen wieder ab. Die spontane Abstrahlung lässt sich aber unterdrücken, wenn intensives Licht einer dieser energieärmeren Wellenlängen zusätzlich eingestrahlt wird: Dann wird die Energie des angeregten Fluorochroms künstlich abgeregt, es kommt zur stimulierten Emission. Von dieser Abregung (engl. Depletion) mittels stimulierter Emission kommt auch die Bezeichnung des Verfahrens.
Bei der STED-Mikroskopie wird ein Laserstrahl für die Anregung der Fluorochrome in das Präparat fokussiert. Gleichzeitig wird in die Außenbereiche des Fokus ein Ring aus Abregungslicht gelegt, sodass spontanes Fluoreszenzlicht nur aus einem zentralen Bereich abgestrahlt wird, der kleiner ist als der beugungsbegrenzte Anregungsfokus. Der STED-Effekt ist zunächst auf eine Stelle im Präparat begrenzt. Diese Stelle wird daher wie bei anderen Laser-Scanning-Mikroskopen über das Präparat gerastert, um zwei- oder dreidimensionale Bilder zu erzeugen.
STED wurde 1986 von Victor Okhonin in einem sowjetischen Patent zum ersten Mal theoretisch beschrieben.[1] 1994 wurde die Idee von Stefan Hell und Jan Wichmann publiziert[2] und 1999 von Stefan Hell und Thomas Klar experimentell realisiert[3]. Das STED-Mikroskop und die Gruppe um Stefan Hell wurden für ihre Ergebnisse im Jahr 2006 mit dem Deutschen Zukunftspreis ausgezeichnet. Im Oktober 2014 wurde Stefan Hell für die Arbeiten am STED mit dem Nobelpreis für Chemie[4][5][6][7] ausgezeichnet. Es wird unter anderem in der Arbeitsgruppe von Stefan Hell am Max-Planck-Institut für biophysikalische Chemie in Göttingen weiterentwickelt. STED-Mikroskope sind auch kommerziell erhältlich.[8]
Grundlagen
Aufgrund von Beugung ist die Auflösung herkömmlicher Lichtmikroskope begrenzt, eine Grenze, die als Abbe-Limit bezeichnet wird: Es lassen sich keine Details auflösen, die kleiner als circa die halbe Wellenlänge des verwendeten Lichts sind. Nebeneinander liegende Strukturen werden nur ab einem Abstand von ca. 200 nm aufgelöst. Die Auflösung für hintereinander liegende Objekte (Tiefenauflösung) ist noch schlechter.
Beim STED-Mikroskop wird dieses Abbe-Limit nicht außer Kraft gesetzt, sondern gezielt „überlistet“: durch Ausschalten von Fluoreszenzfarbstoffen wird der übrigbleibende noch fluoreszierende Bereich stark verkleinert (siehe unten) und damit Auflösung unter der Abbé-Grenze ermöglicht. Es konnte bereits eine Auflösung von 2,4 nm (lateral) erzielt werden.[9]
Ein STED-Mikroskop baut auf Fluoreszenz-Laser-Raster-Mikroskopen auf. Um das Funktionsprinzip besser zu verstehen, ist es notwendig, auf die wichtigsten Gesichtspunkte von Fluoreszenz, stimulierter Emission und der Laser-Raster-Mikroskopie einzugehen:
Fluoreszenz und Stimulierte Emission
Bei der STED-Mikroskopie werden sogenannte Fluoreszenzfarbstoffe zum Markieren einzelner Bereiche eines Präparats eingesetzt. Solche Farbstoffe können durch Licht bestimmter Wellenlängen (Farben) „angeregt“ werden: Sie absorbieren ein Photon und gehen in einen energiereicheren Zustand über. Aus diesem Zustand können sie nach kurzer Zeit spontan durch Aussenden eines Photons größerer Wellenlänge (anderer Farbe) wieder in den Grundzustand zurückkehren. Diese spontane Abstrahlung von Licht nennt man Fluoreszenz. Durch geeignete Farbfilter kann das Fluoreszenzlicht vom Anregungslicht getrennt werden.
Ein angeregtes Farbstoffmolekül kann außer durch Fluoreszenz auch durch stimulierte Emission wieder in den Grundzustand zurückkehren. Dies passiert, wenn das angeregte Farbstoffmolekül mit Licht von ungefähr der gleichen Wellenlänge wie der des Fluoreszenzlichts bestrahlt wird. Das angeregte Farbstoffmolekül kann so zum sofortigen Übergang in den Grundzustand durch Aussenden eines Photons exakt derselben Wellenlänge stimuliert werden. Spontanes Fluoreszenzlicht kann dann nicht mehr ausgesendet werden, denn das Molekül ist nicht mehr im angeregten Zustand. Spontanes Fluoreszenzlicht und Licht von der Stimulierten Emission können z. B. durch Farbfilter voneinander getrennt werden.
Fluoreszenz-Laser-Raster-Mikroskopie
Für die Untersuchung in einem Fluoreszenzmikroskop werden Fluoreszenzfarbstoffe an bestimmte Stellen des zu untersuchenden Präparats gebracht. Wird das Präparat nun mit Licht geeigneter Wellenlänge beleuchtet, so werden die Farbstoffe zur Fluoreszenz angeregt und man erhält ein Bild der Farbstoffverteilung im Präparat. Im Laser-Raster-Mikroskop wird nicht das ganze Präparat auf einmal beleuchtet, sondern ein Laserstrahl („Anregungsstrahl“) wird lediglich auf einen kleinen Punkt des Präparats fokussiert. Die in diesem Bereich übrig bleibende scharfe Fluoreszenz wird detektiert. Durch Bewegen (Scanning, Rastern) des Fokus über das Präparat wird punktweise ein Abbild der gefärbten Bereiche erstellt. Die Größe des Fokus bestimmt die maximale Feinheit der Details, die in dem Präparat gerade noch aufgelöst (getrennt wahrgenommen) werden können. Aufgrund der Beugung kann der Fokus nicht beliebig klein gewählt werden. Ein Laserstrahl lässt sich nicht auf einen Fleck kleiner als circa seine halbe Wellenlänge fokussieren.
Funktionsprinzip des STED-Mikroskops

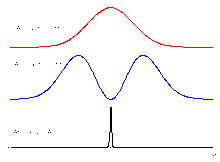
Mit einem STED-Mikroskop ist eine bessere Auflösung als mit einem herkömmlichen Laser-Raster-Mikroskop möglich: Der Bereich, aus dem Fluoreszenz emittiert wird, wird dabei bedeutend kleiner gemacht als der Bereich, der von dem Laserstrahl beleuchtet wird. Das wird durch gezieltes Ausschalten der Farbstoffmoleküle im Außenbereich des Fokus erreicht. Dazu wird das Präparat nicht nur mit dem fokussierten Anregungsstrahl beleuchtet (linkes Bild), sondern gleichzeitig mit einem zweiten Laserstrahl, dem „Ausschaltestrahl“. Diesem Ausschaltestrahl gibt man ein ringförmiges Profil im Fokus (mittleres Bild). In der Mitte, also dort wo der Anregungsstrahl seine maximale Helligkeit hat, ist der Ausschaltestrahl vollkommen dunkel. Der Ausschaltestrahl beeinflusst also die Fluoreszenzfarbstoffe in der Mitte nicht. Er schaltet aber die Fluoreszenzfarbstoffe im Außenbereich des Anregungsfokus durch stimulierte Emission (siehe oben) aus; die Farbstoffmoleküle im Außenbereich bleiben dunkel, obwohl sie von dem Anregungslaser beleuchtet werden. Es leuchten deshalb nur die Farbstoffmoleküle genau aus dem Zentrum (rechtes Bild). Der minimale Durchmesser des Anregungsstrahls ist zwar genauso beugungsbegrenzt wie das zentrale Dunkelfeld des Ausschaltestrahls. Allerdings genügen wenige Photonen des Ausschaltestrahls zur Stimulation der Emission einer größeren Zahl von angeregten Zuständen; außerdem kann die Intensität des Ausschaltestrahls höher als die des Anregungsstrahls gewählt werden. Dadurch ist der nicht ausgeschaltete zentrale Bereich sehr viel kleiner als der mit dem Anregungslaser beleuchtete Bereich (siehe Linienprofile rechts). Beim Scannen des Präparates erfasst man somit jeweils einen leuchtenden Fleck, der viel kleiner ist als in einem normalen Laser-Raster-Mikroskop. Deshalb kann man feinere Details auflösen. Um ein vollständiges Bild zu erhalten, wird das Präparat Punkt für Punkt abgerastert.
Die Größe des resultierenden Lichtflecks sinkt mit steigender Intensität des Ausschaltestrahls immer mehr ab. Das bedeutet, die Auflösung steigt umso weiter an, je heller der Ausschaltestrahl ist; das erreichbare Auflösungsvermögen ist prinzipiell nicht begrenzt.[10] Vor der Erfindung der STED-Mikroskopie bestand das Problem, dass der Anregungsstrahl aufgrund der Abbeschen Beugungsgrenze nicht beliebig klein fokussiert werden kann. Man regt also immer alle Moleküle, die sich gerade im Fokus befinden, an und kann daher nicht entscheiden, von welchem Molekül die Fluoreszenz gerade kommt. Daher konnten Strukturen, die kleiner sind als die Ausdehnung des Laserfokus, nicht unterschieden werden.
Anwendungen
Ein bedeutendes Problem gleich welcher lichtmikroskopischen Technik ist der mangelnde Kontrast von Zellbestandteilen. Schon lange benutzt man deshalb fluoreszente Moleküle, die z. B. mit gentechnischen Methoden oder mittels Antikörpern selektiv an bestimmte Moleküle einer Zelle geheftet werden können. Man kann zum Beispiel Farbstoffe nur an Mitochondrien anbauen. Beleuchtet man nun eine Stelle der so präparierten Zelle mit einem fokussierten Laserstrahl und erhält von dort Fluoreszenz, so waren an genau dieser Stelle Farbstoffmoleküle und damit auch Mitochondrien. Um ein vollständiges Bild zu erhalten, wird das Präparat Punkt für Punkt abgerastert. In einem STED-Mikroskop lassen sich alle Präparate untersuchen, die mit Fluoreszenzfarbstoffen markierbar sind. Anders als bei Elektronenmikroskopen sind kein Vakuum und keine dünnen Schnitte erforderlich. Deshalb lassen sich auch lebende Zellen beobachten[11].
Im Gegensatz zur Rastersondenmikroskopie, einem Nahfeld-Verfahren, ist die STED-Mikroskopie eine Fernfeld-Technik. Sie ist also nicht auf die Untersuchung von Oberflächen beschränkt. Man kann beispielsweise auch das Innere von Zellen untersuchen. Auch die Beobachtung von schnellen dynamischen Prozessen ist möglich[12], mit bis zu 200 Bildern pro Sekunde[13].
Literatur
- Marcus Dyba, Stefan W. Hell: Focal spots of size lambda/23 open up far-field florescence microscopy at 33 nm axial resolution. In: Physical Review Letters. Vol. 88, Nr. 16, 2002, S. 163901, doi:10.1103/PhysRevLett.88.163901.
- Katrin I. Willig, Silvio O. Rizzoli, Volker Westphal, Reinhard Jahn, Stefan W. Hell: STED-microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. In: Nature. Vol. 440, 2005, S. 935–939, doi:10.1038/nature04592.
- Stefan W. Hell: Microscopy and its focal switch. In: Nature Methods. Vol. 6, Nr. 1, 2009, S. 24–32, doi:10.1038/nmeth.1291.
- Stefan W. Hell: Far-Field Optical Nanoscopy. In: Science. Vol. 316, 2007, S. 1153–1158, doi:10.1126/science.1137395.
Weblinks
- Abteilung NanoBiophotonics am Max-Planck-Institut für biophysikalische Chemie (englisch)
- Detailinformationen und Bilder zur STED-Mikroskopie (PDF-Datei; 426 kB)
- Deutscher Zukunftspreis 2006: Lichtmikroskopie in ungekannter Schärfe – Hintergrundmaterial
- STED – Mikroskopie jenseits optischer Grenzen, Film im Youtube-Kanal der Max-Planck-Gesellschaft
Einzelnachweise
- Patent SU1374922A1: Способ исследования микроструктуры образца. Angemeldet am 10. April 1986, veröffentlicht am 30. Juli 1991, Anmelder: Институт Биофизики CO АН СССР, Erfinder: В. А. Охонин.
- Stefan W. Hell and Jan Wichmann: Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. In: Optics Letters. Band 19, Nr. 11, 1994, S. 780–782, doi:10.1364/OL.19.000780.
- Thomas A. Klar, Stefan W. Hell: Subdiffraction resolution in far-field fluorescence microscopy. In: Optics Letters. Vol. 24, Nr. 14, 1999, S. 954–956, doi:10.1364/OL.24.000954.
- The Nobel Prize in Chemistry 2014. In: NobelPrize.org. Nobel Media AB, 2014, abgerufen am 13. Oktober 2020 (englisch).
- Holger Dambeck: Messerscharfer Blick ins Innerste des Lebens. Chemie-Nobelpreis 2014. In: Spiegel Online. Rudolf Augstein, 9. Oktober 2012, abgerufen am 9. Oktober 2012.
- Norbert Lossau: Nobelpreis für die Entwickler des Supermikroskops. In: Welt. Stefan Aust, 8. Oktober 2014, abgerufen am 9. Oktober 2014.
- Chemie-Nobelpreis 2014 geht an Max-Planck-Forscher Stefan Hell. Max-Planck-Institut für biophysikalische Chemie, 8. Oktober 2014, abgerufen am 9. Oktober 2014.
- Geräte von PicoQuant siehe MicroTime 200 STED, Geräte von Abberior Instruments siehe INFINITY, Geräte von Leica Microsystems siehe TCS STED.
- D. Wildanger, B. R. Patton, H. Schill, L. Marseglia, J. P. Hadden, S. Knauer, A. Schönle, J. G. Rarity, J. L. O’Brien, S. W. Hell, J. M. Smith:: Solid Immersion Facilitates Fluorescence Microscopy with Nanometer Resolution and Sub-Ångström Emitter Localization. In: Advanced Materials. 2012, doi:10.1002/adma.201203033.
- Stefan W. Hell: Far-Field Optical Nanoscopy. In: Science. Vol. 316, 2007, S. 1153–1158, doi:10.1126/science.1137395.
- Volker Westphal, Silvio O. Rizzoli, Marcel A. Lauterbach, Dirk Kamin, Reinhard Jahn, Stefan W. Hell: Video-Rate Far-FieldOptical Nanoscopy Dissects Synaptic Vesicle Movement. In: Science. Vol. 320, 2008, ISSN 0036-8075, S. 246–249, doi:10.1126/science.1154228.
- Volker Westphal, Marcel A. Lauterbach, Angelo Di Nicola, Stefan W. Hell: Dynamic far-field fluorescence nanoscopy. In: New Journal of Physics. Band 9, 2007, S. 435 ff., doi:10.1088/1367-2630/9/12/435.
- Marcel A. Lauterbach, Chaitanya K. Ullal, Volker Westphal, and Stefan W. Hell: Dynamic Imaging of Colloidal-Crystal Nanostructures at 200 Frames per Second. In: Langmuir. Band 26, 2010, S. 14400–14404., doi:10.1021/la102474p.