DNA-Sequenzierung
DNA-Sequenzierung ist die Bestimmung der Nukleotid-Abfolge in einem DNA-Molekül. Die DNA-Sequenzierung hat die biologischen Wissenschaften revolutioniert und die Ära der Genomik eingeleitet. Seit 1995 konnte durch DNA-Sequenzierung das Genom von über 50.000 (Stand: 2020) verschiedenen Organismen analysiert werden. Zusammen mit anderen DNA-analytischen Verfahren wird die DNA-Sequenzierung u. a. auch zur Untersuchung genetisch bedingter Erkrankungen herangezogen. Darüber hinaus ist die DNA-Sequenzierung als analytische Schlüsselmethode, insbesondere im Rahmen von DNA-Klonierungen (engl. molecular cloning), aus einem molekularbiologischen bzw. gentechnischen Laborbetrieb heute nicht mehr wegzudenken.
Problemstellung
Die DNA-Sequenzierung als Ablesen der Nukleotidfolge von DNA war über Jahrzehnte hinweg bis in die Mitte der 1970er Jahre ein ungelöstes Problem, bis entsprechende biochemische bzw. biotechnologische Methoden entwickelt wurden. Heutzutage ist selbst die Sequenzierung ganzer Genome vergleichsweise schnell und einfach geworden.[1]
Die Herausforderungen einer Genomsequenzierung beschränken sich jedoch nicht nur auf das direkte Ablesen der Nukleotidsequenz. Je nach Verfahren werden in jeder einzelnen Sequenzierreaktion auf Grund technischer Beschränkungen nur kurze DNA-Abschnitte (engl. reads) bis maximal 1000 Basenpaare abgelesen. Nach Erhalt der Sequenz wird dann der nächste Primer (mit einer Sequenz aus dem Ende der vorigen Sequenzierung) hergestellt, was als Primer Walking oder bei ganzen Chromosomen als chromosome walking bezeichnet wird und 1979 erstmals angewendet wurde.[2]
Ein größeres Sequenzierprojekt, wie das Humangenomprojekt, bei dem mehrere Milliarden Basenpaare sequenziert wurden, erfordert darum eine Herangehensweise, die als Shotgun Sequencing bezeichnet wird. Dabei werden längere DNA-Abschnitte zunächst in kleinere Einheiten zerlegt, diese dann sequenziert und die Sequenzinformationen der einzelnen kurzen Abschnitte anschließend mit bioinformatischen Methoden wieder zu einer vollständigen Gesamtsequenz zusammengefügt. Um aus den rohen Sequenzdaten biologisch relevante Informationen zu gewinnen (beispielsweise Informationen über vorhandene Gene und deren Kontrollelemente), schließt sich an die Sequenzierung die DNA-Sequenzanalyse an. Ohne sie bleibt jede Sequenzinformation ohne wissenschaftlichen Wert.
Sequenzierungsmethoden

Es gibt heute mehrere Verfahren zum Ablesen der Sequenzinformation von einem DNA-Molekül. Lange Zeit waren überwiegend Weiterentwicklungen der Methode nach Frederick Sanger in Verwendung. Moderne Verfahren bieten Möglichkeiten der beschleunigten Sequenzierung durch hochparallelen Einsatz. Die nach der Sanger-Methode entwickelten Sequenzierungsverfahren werden häufig als Sequenzierung der nächsten Generation (engl. next generation sequencing) bezeichnet.
Methode von Maxam und Gilbert
Die Methode von Allan Maxam und Walter Gilbert von 1977 beruht auf der basenspezifischen chemischen Spaltung der DNA durch geeignete Reagenzien und anschließender Auftrennung der Fragmente durch denaturierende Polyacrylamid-Gelelektrophorese.[3] Die DNA wird zunächst an einem 5' oder 3'-Ende mit radioaktivem Phosphat oder einem nicht-radioaktiven Stoff (Biotin, Fluorescein) markiert. In vier getrennten Ansätzen werden dann jeweils bestimmte Basen vom Zucker-Phosphat-Rückgrat der DNA partiell (limitiert) modifiziert und abgespalten, beispielsweise wird die Base Guanin (G) durch das Reagenz Dimethylsulfat methyliert und durch Alkalibehandlung mit Piperidin entfernt. Danach wird der DNA-Strang komplett gespalten. In jedem Ansatz entstehen Fragmente unterschiedlicher Länge, deren 3'-Ende stets an bestimmten Basen gespalten worden war. Die denaturierende Polyacrylamid-Gelelektrophorese trennt die Fragmente nach der Länge auf, wobei Längenunterschiede von einer Base aufgelöst werden. Durch Vergleich der vier Ansätze auf dem Gel lässt sich die Sequenz der DNA ablesen. Ihren Erfindern ermöglichte diese Methode die Bestimmung der Operon-Sequenz eines Bakteriengenoms. Die Methode kommt heute kaum noch zum Einsatz, da sie gefährliche Reagenzien benötigt und schwerer automatisierbar ist als die zur gleichen Zeit entwickelte Didesoxymethode nach Sanger.
Didesoxymethode nach Sanger
Die Didesoxymethode nach Sanger wird auch Kettenabbruch-Synthese genannt. Sie stellt eine enzymatische Methode dar. Sie wurde von Sanger und Coulson um 1975 entwickelt und bereits 1977 mit der ersten vollständigen Sequenzierung eines Genoms (Bakteriophage φX174)[4] vorgestellt.[5] Sanger erhielt für seine Arbeiten zur DNA-Sequenzierung zusammen mit Walter Gilbert und Paul Berg 1980 den Nobelpreis für Chemie.[6]
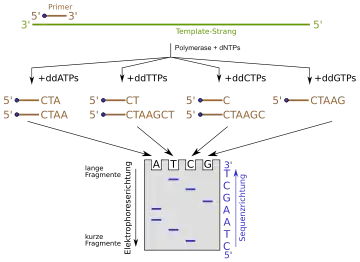
dNTP ist die allgemeine Abkürzung für ein Nukleosidtriphosphat und kann für dATP, dCTP, dGTP oder dTTP stehen. ddNTPs sind die entsprechenden Didesoxy-Varianten der dNTPs. Der Einbau eines ddNTPs führt zum Abbruch der Polymerisationsreaktion. Die blauen Punkte am 5'-Ende des Primers stellen eine Markierung dar (z. B. eine fluoreszierende Gruppe), mittels der die Syntheseprodukte später im Gel sichtbar gemacht werden können. Alternativ lassen sich auch radioaktiv markierte Nukleosidtriphosphate zur Polymerisationsreaktion einsetzen.
Ausgehend von einem kurzen Abschnitt bekannter Sequenz (Primer) wird durch eine DNA-Polymerase einer der beiden komplementären DNA-Stränge verlängert. Zunächst wird die DNA-Doppelhelix durch Erwärmung denaturiert, woraufhin Einzelstränge für das weitere Vorgehen zur Verfügung stehen. In vier sonst gleichen Ansätzen (alle beinhalten die vier Nukleotide, von denen eines radioaktiv markiert ist[7]) wird je eine der vier Basen zum Teil als Didesoxynukleosidtriphosphat (ddNTP) zugegeben (also je ein Ansatz mit entweder ddATP, ddCTP, ddGTP oder ddTTP). Diese Kettenabbruch-ddNTPs besitzen keine 3'-Hydroxygruppe: Werden sie in den neusynthetisierten Strang eingebaut, ist eine Verlängerung der DNA durch die DNA-Polymerase nicht mehr möglich, da die OH-Gruppe am 3'-C-Atom für die Verknüpfung mit der Phosphatgruppe des nächsten Nukleotids fehlt. In der Folge entstehen DNA-Fragmente unterschiedlicher Länge, die in jedem einzelnen Ansatz stets mit dem gleichen ddNTP enden (also je Ansatz nur mit A oder C oder G oder T). Nach der Sequenzier-Reaktion werden die markierten Abbruchprodukte aus allen Ansätzen mittels Polyacrylamid-Gelelektrophorese der Länge nach aufgetrennt. Durch Vergleich der vier Ansätze kann man die Sequenz nach der Exposition des radioaktiven Gels auf einem fotografischen Film (Röntgenfilm) ablesen. Die dementsprechend komplementäre Sequenz ist die Sequenz der verwendeten einsträngigen DNA-Matrize. Als Sequenzier-Reaktion kommt heutzutage eine Variation der Polymerase-Kettenreaktion (PCR) zum Einsatz. Anders als bei der PCR wird nur ein Primer eingesetzt, sodass die DNA nur linear amplifiziert wird.
Ein radioaktives Verfahren zur DNA-Sequenzierung, durch den Transfer der DNA Moleküle auf einen Träger während der elektrophoretischen Auftrennung, wurde von Fritz M. Pohl und seiner Arbeitsgruppe Anfang der 1980er Jahre entwickelt.[8][9] Die Vermarktung des „Direct-Blotting-Electrophoresis System GATC 1500“, erfolgte durch das Konstanzer Unternehmen GATC Biotech. Der DNA-Sequenzierer wurde z. B. im Rahmen des europäischen Genomprojekts zur Sequenzierung des Chromosoms II der Hefe Saccharomyces cerevisiae eingesetzt.[10]
Seit Anfang der 1990er Jahre werden vor allem mit Fluoreszenz-Farbstoffen markierte Didesoxynukleosidtriphosphate eingesetzt. Jedes der vier ddNTPs wird mit einem unterschiedlichen Farbstoff gekoppelt. Diese Modifikation erlaubt es, alle vier ddNTPs in einem Reaktionsgefäß zuzugeben, eine Aufspaltung in getrennte Ansätze und der Umgang mit Radioisotopen entfällt. Die entstehenden Kettenabbruchprodukte werden mittels Kapillarelektrophorese aufgetrennt und mit Hilfe eines Lasers zur Fluoreszenz angeregt. Die ddNTPs am Ende jedes DNA-Fragmentes zeigen dadurch Fluoreszenz unterschiedlicher Farbe und können so von einem Detektor erkannt werden. Das Elektropherogramm (die Abfolge der Farbsignale, die am Detektor erscheinen) gibt direkt die Sequenz der Basen des sequenzierten DNA-Stranges wieder.
Moderne Ansätze
Mit der zunehmenden Bedeutung der DNA-Sequenzierung in der Forschung und Diagnostik wurden Methoden entwickelt, die einen erhöhten Durchsatz erlauben. Damit ist es nun möglich, das komplette menschliche Genom in etwa 8 Tagen zu sequenzieren.[11] Die entsprechenden Verfahren werden als Sequenzierung der zweiten Generation (engl. second generation sequencing) bezeichnet. Verschiedene Firmen haben Verfahren mit unterschiedlichen Vor- und Nachteilen entwickelt. Außer den hier aufgeführten gibt es noch weitere. Die DNA-Sequenzierung der zweiten Generation wurde von der Zeitschrift Nature Methods zur Methode des Jahres 2007 gekürt.[12]
Pyrosequenzierung
Die Pyrosequenzierung nutzt wie die Sanger-Sequenzierung eine DNA-Polymerase zur Synthese des DNA-Gegenstranges,[13] wobei der Typ der DNA-Polymerase durchaus noch unterschiedlich sein kann. Die DNA-Mischung wird mit einem DNA-Adapter ligiert und über eine komplementäre Adaptersequenz an beads gekoppelt. Die mit DNA beladenen beads werden auf eine Platte mit Poren von der Größe eines beads gegeben, bei der unter jeder Pore ein Lichtleiter zu einem Detektor führt. Die DNA-Polymerase wird gewissermaßen „in Aktion“ beobachtet, wie sie nacheinander einzelne Nukleotide an einen neusynthetisierten DNA-Strang anhängt. Der erfolgreiche Einbau eines Nukleotids wird durch ein ausgeklügeltes Enzymsystem unter Beteiligung einer Luziferase in einen Lichtblitz umgesetzt und von einem Detektor erfasst. Die zu sequenzierende DNA dient als Matrizenstrang und liegt einzelsträngig vor. Ausgehend von einem Primer erfolgt die Strangverlängerung, Nukleotid um Nukleotid, durch Zugabe von jeweils einer der vier Arten der Desoxynukleosidtriphosphate (dNTP). Bei Zugabe des passenden (komplementären) Nukleotids erhält man ein Signal. Wurde ein an dieser Stelle nicht passendes NTP zugegeben, bleibt der Lichtblitz aus. Danach werden die vorhandenen NTP zerstört, und eine andere Art wird zugesetzt; dies wird fortgesetzt, bis sich wieder eine Reaktion zeigt; spätestens nach der vierten Zugabe zeigt sich eine Reaktion, da dann alle Arten von NTP durchprobiert wurden.
Bei Einbau eines komplementären Nukleotids durch die DNA-Polymerase wird Pyrophosphat (PPi) freigesetzt. Das Pyrophosphat wird durch die ATP-Sulfurylase zu Adenosintriphosphat (ATP) umgesetzt. Das ATP treibt die Luziferase-Reaktion an, wodurch Luziferin in Oxyluziferin umgesetzt wird. Dies resultiert wiederum in einem detektierbaren Lichtsignal – dessen Stärke proportional zum verbrauchten ATP ist.
Die Pyrosequenzierung wird beispielsweise zur Bestimmung der Häufigkeit von bestimmten Genmutationen (SNPs, engl. Single Nucleotide Polymorphism), z. B. bei der Untersuchung von Erbkrankheiten eingesetzt. Die Pyrosequenzierung ist gut automatisierbar und eignet sich zur hochparallelen Analyse von DNA-Proben.
Pyrosequenzierung wurde Mitte der 1990er Jahre von Mathias Uhlén, Mostafa Ronaghi und Pål Nyrén entwickelt (Nyrén erhielt dafür 2013 den Europäischen Erfinderpreis) und ab 1999 von Jonathan Rothberg in der 454 GS FLX der Firma 454 Life Sciences mit Chip-Technologie umgesetzt (siehe Ionen-Halbleiter-DNA-Sequenzierungssystem), die 2005 als erste Next Generation Plattform auf den Markt kam (aufgekauft von Roche Diagnostics 2007).[14] Mit der 454 GS FLX gelang es 2007 das Genom von James Watson, der die Doppelhelixstruktur der DNA 1953 mit Francis Crick entdeckte, in nur 2 Monaten zu sequenzieren, während das 2003 abgeschlossene erste Human Genome Project noch 13 Jahren benötigte.[15]
Sequenzierung durch Hybridisierung
Zu diesem Zweck werden auf einem Glasträger (DNA-Chip oder Microarray) kurze DNA-Abschnitte (Oligonukleotide) in Reihen und Spalten fixiert. Die Fragmente der zu sequenzierenden DNA werden mit Farbstoffen markiert und das Fragmentgemisch wird auf der Oligonukleotidmatrix aufgebracht, so dass komplementäre fixierte und freie DNA-Abschnitte miteinander hybridisieren können. Nach dem Auswaschen ungebundener Fragmente lässt sich das Hybridisierungsmuster anhand der Farbmarkierungen und deren Stärke ablesen. Da die Sequenzen der fixierten Oligonukleotide und deren Überlappungsbereiche bekannt sind, kann man letztlich aus dem Farbmuster auf die zugrundeliegende Gesamtsequenz der unbekannten DNA rückschließen.
Ionen-Halbleiter-DNA-Sequenzierungssystem
Dieses Verfahren von Ion Torrent nutzt Halbleiterverfahren, um mittels integrierter Schaltkreise eine unmittelbare nicht-optische Genom-Sequenzierung durchzuführen. Dabei werden die Sequenzierungsdaten direkt über die Halbleiterchip-Detektion von Ionen gewonnen, die von vorlageabhängigen DNA-Polymerasen produziert werden. Der dafür verwendete Chip besitzt ionensensitive Feldeffekttransistor-Sensoren, die in einem Raster von 1,2 Mio. Vertiefungen angeordnet sind, in denen die Polymerase-Reaktion stattfindet. Dieses Raster ermöglicht parallele und simultane Detektion unabhängiger Sequenzreaktionen. Dabei kommt die komplementäre Metalloxid-Halbleiter-Technologie (CMOS) zum Einsatz, die eine kostengünstige Reaktion in hoher Messpunkt-Dichte erlaubt.[16]
Ein erster solcher Chip wurde von Jonathan Rothberg entwickelt für die erste Next-Generation-Plattform, die 454 GS FlX, die Pyrosequenzierung benutzt (siehe oben).
Sequenzierung mit Brückensynthese
Die zu sequenzierende doppelsträngige DNA wird bei der Sequenzierung mit Brückensynthese von Solexa/Illumina an beiden Enden mit je einer unterschiedlichen Adapter-DNA-Sequenz ligiert. Anschließend wird die DNA denaturiert, nach Verdünnung einzelsträngig auf eine Trägerplatte ligiert und per Brückenamplifikation in situ vervielfältigt.[17][18] Dadurch entstehen auf der Trägerplatte einzelne Bereiche (cluster) mit vervielfältigter DNA, die innerhalb eines clusters die gleiche Sequenz aufweisen. In einer sequencing by synthesis-verwandten PCR-Reaktion mit vier verschiedenfarbig fluoreszierenden Kettenabbruchsubstraten wird in Echtzeit die jeweils eingebaute Nukleinbase pro Zyklus in einem cluster bestimmt. Die Methode wurde 1997 durch Shankar Balasubramanian und David Klenerman entwickelt, die dafür für 2022 den Breakthrough Prize in Life Sciences erhielten. Die Methode wurde im Rahmen des Startups Solexa entwickelt, die 2006 von Illumina übernommen wurden, daher der Name. In die Solexa/Illumina-Methode flossen damals auch die unabhängig in Genf bei GlaxoSmithKline erstellten Pionierarbeiten von Pascal Mayer ein (insbesondere die Cluster-Vervielfältigung), der dafür ebenfalls den Breakthrough Prize erhielt. Die Plattform von Solexa kam 2007 auf den Markt und machte später Illumina zum Marktführer.
Zwei-Basen-Sequenzierung
Die Zwei-Basen-Sequenzierung (engl. Sequencing by Oligo Ligation Detection, SOLiD) von Applied Biosystems ist eine Variante der Sequencing by Ligation.[18][19] Eine DNA-Bibliothek wird verdünnt und mit einer DNA-Polymerase an microbeads gekoppelt, anschließend werden in einer Emulsions-PCR die DNA vervielfältigt. Dadurch enthält jedes Microbead mehrere Kopien jeweils nur einer DNA-Sequenz. Die microbeads werden am 3'-Ende modifiziert, wodurch sie einzeln auf einer Trägerplatte befestigt werden können. Nach Bindung von Primern und einer Zugabe von vier verschiedenen spaltbaren Sonden, die jeweils farblich unterschiedlich fluoreszent markiert sind und anhand der ersten beiden Nukleotide (CA, CT, GG, GC) an die DNA-Vorlage binden, wird mit einer DNA-Ligase ligiert. Anschließend werden die Sonden gespalten, wodurch die Markierungen freigesetzt werden. Durch bis zu fünf Primer, die jeweils in der Sequenz um eine Base zurückversetzt sind, wird jede Base in der DNA-Sequenz in mindestens zwei verschiedenen Ligationsreaktionen bestimmt.
Die Methode (die einzige der frühen Plattformen, die Ligasen benutzten), wurde von Kevin McKernan bei Applied Biosystems entwickelt und kam im Oktober 2007 als SOLiD-Plattform auf den Markt. Die Ligationsmethode wurde zuvor bei 2006 von Applied Biosystems übernommenen Firma Agencourt Personal Genomics entwickelt.[14]
Sequenzierung mit gepaarten Enden
Ein eindeutig identifizierbares Signal erhält man auch über die Erzeugung von kurzen DNA-Stücken aus dem Anfang und Ende einer DNA-Sequenz (engl. Paired End Tag Sequencing, PETS), wenn das Genom bereits vollständig sequenziert wurde.[20][21]
Sequenzierung der dritten Generation
Die Sequenzierung der dritten Generation misst erstmals die Reaktion bei einzelnen Molekülen als Einzelmolekülexperiment, wodurch eine der Sequenzierung vorangehende Amplifikation per PCR entfällt.[22][23] Dadurch wird die ungleichmäßige Amplifikation durch thermostabile DNA-Polymerasen vermieden, da Polymerasen manche DNA-Sequenzen bevorzugt binden und diese verstärkt replizieren (engl. polymerase bias). Dadurch können manche Sequenzen übersehen werden. Weiterhin kann das Genom einzelner Zellen untersucht werden. Die Aufnahme des freigesetzten Signals wird in Echtzeit aufgenommen. Bei der DNA-Sequenzierung der dritten Generation werden, je nach Verfahren, zwei verschiedene Signale aufgezeichnet: Freigesetzte Protonen (als Variante der Halbleitersequenzierung) oder Fluorophore (mit Fluoreszenzdetektor).[18] Die DNA- und RNA-Sequenzierung einzelner Zellen wurde von der Zeitschrift Nature Methods zur Methode des Jahres 2013 gekürt.[24]
Nanoporen-Sequenzierung
Die Nanoporen-Sequenzierung beruht auf Änderungen des Ionenstromes durch Nanoporen, die in eine künstlich erzeugte Membran eingelagert sind. Als Nanopore werden sowohl biologische (kleine Transmembranproteine ähnlich einem Ionenkanal, z. B. α-Hämolysin (α-HL) oder ClpX)[25] als auch synthetische Poren (aus Siliciumnitrid oder Graphen) sowie halbsynthetische Poren verwendet. Die Nanopore ist eingelassen in eine künstliche Membran, die einen besonders hohen elektrischen Widerstand aufweist. Die Pore ist im Gegensatz zu gewöhnlichen Ionenkanälen permanent geöffnet und erlaubt somit, nach Anlegung eines Potentials, einen konstanten Ionenfluss durch die Membran. DNA-Moleküle, die die Pore passieren, führen zu einer Verringerung des Stromes. Diese Stromabnahme hat eine für jedes Nukleotid spezifische Amplitude, welche gemessen und dem entsprechenden Nukleotid zugeordnet werden kann. Bei der Einzelstrangsequenzierung wird ein doppelsträngiger DNA-Strang durch eine Helikase getrennt und in die Nanopore eingeführt. Im Falle einer MspA-Pore befinden sich gleichzeitig vier Nukleotide der DNA innerhalb der Pore.[26] Die Durchtrittsgeschwindigkeit ist unter anderem von der pH-Wert-Differenz beidseitig der Membran abhängig.[27] Durch die spezifischen Ionenstromänderungen für jedes der vier Nukleotide lässt sich aus dem erhaltenen Datensatz die Sequenz ablesen. Eine Auswertung erfolgt z. B. mit der Software Poretools.[28] Der Vorteil dieser Methode besteht darin, dass sie auch bei langen DNA-Strängen eine gleichbleibende Genauigkeit aufweist. Eine Abwandlung der Methode wird zur Proteinsequenzierung verwendet.[29]
Die Nanoporen-Sequenzierungs-Technologie wird beispielsweise durch die britische Firma Oxford Nanopore Technologies vorangetrieben. Deren „MinION“-Sequenzierer war anfänglich nur über ein sogenanntes „Early Access Programme“ zugänglich,[30] ist jedoch seit 2015 über herkömmliche Vertriebswege zu erwerben.
Literatur
- Laura Bonetta: Genome sequencing in the fast lane. (PDF; 751 kB) In: Nature Methods, Band 3, 2006, S. 141–147. doi:10.1038/nmeth0206-141
- Karin Hollricher: Hochleistungs-Sequenzieren. In: Laborjournal, Nr. 4, 2009, S. 44–48.
- B. A. Peters, B. G. Kermani et al.: Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. In: Nature, Band 487, Nummer 7406, Juli 2012, S. 190–195. doi:10.1038/nature11236. PMID 22785314. PMC 3397394 (freier Volltext).
- M. W. Schmitt, S. R. Kennedy et al..: Detection of ultra-rare mutations by next-generation sequencing. In: Proceedings of the National Academy of Sciences. Band 109, Nummer 36, September 2012, S. 14508–14513. doi:10.1073/pnas.1208715109. PMID 22853953. PMC 3437896 (freier Volltext).
Weblinks
- Erbmaterial von Erregern vergleichen, um Krankheitsausbrüche aufzuklären. (PDF; 272 kB) Stellungnahme des Bundesinstituts für Risikobewertung (BfR)
Einzelnachweise
- E. Pettersson, J. Lundeberg, A. Ahmadian: Generations of sequencing technologies. In: Genomics. Band 93, Nr. 2, Februar 2009, S. 105–111, doi:10.1016/j.ygeno.2008.10.003, PMID 18992322.
- A. C. Chinault, J. Carbon: Overlap hybridization screening: isolation and characterization of overlapping DNA fragments surrounding the leu2 gene on yeast chromosome III. In: Gene. Band 5(2), 1979, S. 111–126. PMID 376402.
- A. Maxam, W. Gilbert: A new method of sequencing DNA. In: Proceedings of the National Academy of Sciences U.S.A. Band 74, 1977, S. 560–564. PMID 265521; PMC 392330 (freier Volltext).
- F. Sanger et al.: Nucleotide sequence of bacteriophage phi X174 DNA. In: Nature. Band 265, 1977, S. 687–695. doi:10.1038/265687a0;PMID 870828.
- F. Sanger et al.: DNA sequencing with chain-terminating inhibitors. In: Proceedings of the National Academy of Sciences U.S.A. Band 74, 1977, S. 5463–5467. PMID 271968; PMC 431765 (freier Volltext).
- Informationen der Nobelstiftung zur Preisverleihung 1980 an Walter Gilbert, Paul Berg und Frederick Sanger (englisch)
- F. Sanger, A. R. Coulson: A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. In: Journal of Molecular Biology. Band 93, 1975, S. 441–448. PMID 1100841.
- S. Beck, F. M. Pohl: DNA sequencing with direct blotting electrophoresis. In: The EMBO journal. Band 3, Nummer 12, Dezember 1984, S. 2905–2909. PMID 6396083. PMC 557787 (freier Volltext).
- Patentschrift DE3022527.
- H. Feldmann, M. Aigle et al..: Complete DNA sequence of yeast chromosome II. In: The EMBO journal. Band 13, Nummer 24, Dezember 1994, S. 5795–5809, PMID 7813418. PMC 395553 (freier Volltext).
- Genome sequencing: the third generation. 6. Februar 2009, abgerufen am 28. Februar 2011 (englisch).
- Anonym: Method of the Year. In: Nature Methods. 5, 2008, S. 1, doi:10.1038/nmeth1153.
- M. Ronaghi: Pyrosequencing sheds light on DNA sequencing. In: Genome Research. 11, 2001, S. 3–11. PMID 11156611 doi:10.1101/gr.11.1.3 (PDF) (PDF; 685 kB).
- Kelly Rae Chi, The year of sequencing, Nature Methods, Band 5, Januar 2008, S. 11–14.
- Pål Nyrén, inventor of Pyrosequencing, Europäisches Patentamt 2013.
- J. M. Rothberg, W. Hinz, T. M. Rearick, J. Schultz, W. Mileski, M. Davey, J. H. Leamon, K. Johnson, M. J. Milgrew, M. Edwards, J. Hoon, J. F. Simons, D. Marran, J. W. Myers, J. F. Davidson, A. Branting, J. R. Nobile, B. P. Puc, D. Light, T. A. Clark, M. Huber, J. T. Branciforte, I. B. Stoner, S. E. Cawley, M. Lyons, Y. Fu, N. Homer, M. Sedova, X. Miao, B. Reed, J. Sabina, E. Feierstein, M. Schorn, M. Alanjary, E. Dimalanta, D. Dressman, R. Kasinskas, T. Sokolsky, J. A. Fidanza, E. Namsaraev, K. J. McKernan, A. Williams, G. T. Roth, J. Bustillo: An integrated semiconductor device enabling non-optical genome sequencing. In: Nature. 475(7356), 20. Jul 2011, S. 348–352, doi:10.1038/nature10242.
- E. R. Mardis: The impact of next-generation sequencing technology on genetics. In: Trends Genet. Band 24(3), 2008, S. 133–141. doi:10.1016/j.tig.2007.12.007. PMID 18262675.
- L. Liu, Y. Li, S. Li, N. Hu, Y. He, R. Pong, D. Lin, L. Lu, M. Law: Comparison of next-generation sequencing systems. In: J Biomed Biotechnol. Band 2012, S. 251364. doi:10.1155/2012/251364. PMID 22829749; PMC 3398667 (freier Volltext).
- J. Henson, G. Tischler, Z. Ning: Next-generation sequencing and large genome assemblies. In: Pharmacogenomics. Band 13(8), 2012, S. 901–915. doi:10.2217/pgs.12.72. PMID 22676195. (PDF).
- X. Ruan, Y. Ruan: Genome wide full-length transcript analysis using 5' and 3' paired-end-tag next generation sequencing (RNA-PET). In: Methods Mol Biol. Band 809, 2012, S. 535–562. doi:10.1007/978-1-61779-376-9_35. PMID 22113299.
- M. J. Fullwood, C. L. Wei, E. T. Liu, Y. Ruan: Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. In: Genome Res. Band 19(4), 2009, S. 521–532. doi:10.1101/gr.074906.107. PMID 19339662. (PDF).
- F. Ozsolak: Third-generation sequencing techniques and applications to drug discovery. In: Expert Opin Drug Discov. Band 7(3), 2012, S. 231–243. doi:10.1517/17460441.2012.660145. PMID 22468954; PMC 3319653 (freier Volltext).
- C. S. Pareek, R. Smoczynski, A. Tretyn: Sequencing technologies and genome sequencing. In: J Appl Genet. Band 52, Ausgabe 4, 2011, S. 413–435. doi:10.1007/s13353-011-0057-x. PMID 21698376; PMC 3189340 (freier Volltext).
- Method of the Year 2013. In: Nature Methods. 11, 2013, S. 1, doi:10.1038/nmeth.2801.
- J. Nivala, D. B. Marks, M. Akeson: Unfoldase-mediated protein translocation through an α-hemolysin nanopore. In: Nature Biotechnology. Band 31, Nummer 3, März 2013, S. 247–250, doi:10.1038/nbt.2503. PMID 23376966. PMC 3772521 (freier Volltext).
- A. H. Laszlo, I. M. Derrington, B. C. Ross, H. Brinkerhoff, A. Adey, I. C. Nova, J. M. Craig, K. W. Langford, J. M. Samson, R. Daza, K. Doering, J. Shendure, J. H. Gundlach: Decoding long nanopore sequencing reads of natural DNA. In: Nature Biotechnology. Band 32, Nummer 8, August 2014, S. 829–833, doi:10.1038/nbt.2950. PMID 24964173. PMC 4126851 (freier Volltext).
- B. N. Anderson, M. Muthukumar, A. Meller: pH tuning of DNA translocation time through organically functionalized nanopores. In: ACS Nano. Band 7, Nummer 2, Februar 2013, S. 1408–1414, doi:10.1021/nn3051677. PMID 23259840. PMC 3584232 (freier Volltext).
- N. J. Loman, A. R. Quinlan: Poretools: a toolkit for analyzing nanopore sequence data. In: Bioinformatics. Band 30, Nummer 23, Dezember 2014, S. 3399–3401, doi:10.1093/bioinformatics/btu555. PMID 25143291.
- Y. Yang, R. Liu, H. Xie, Y. Hui, R. Jiao, Y. Gong, Y. Zhang: Advances in nanopore sequencing technology. In: Journal of nanoscience and nanotechnology. Band 13, Nummer 7, Juli 2013, S. 4521–4538. PMID 23901471.
- Start using MinION (Memento vom 23. Januar 2016 im Internet Archive), abgerufen am 23. März 2016.