Kuru (Krankheit)
Bei Kuru handelt es sich um eine Prionenkrankheit, die im 20. Jahrhundert epidemieartig beim Volk der Fore in Papua-Neuguinea und in geringerem Ausmaß bei einigen Nachbarvölkern auftrat. Das Wort Kuru stammt aus der Sprache der einheimischen Bevölkerung und bedeutet Muskelzittern.
| Klassifikation nach ICD-10 | |
|---|---|
| A81.8 | Kuru |
| ICD-10 online (WHO-Version 2019) | |
Krankheitszeichen
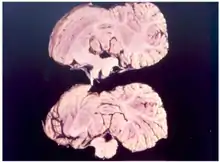
Die Krankheit äußert sich vor allem in Bewegungsstörungen und führt typischerweise innerhalb von 6 bis 12 Monaten nach Auftreten der ersten Symptome zum Tod. Im Einzelnen handelt es sich bei den Symptomen um Gang- und Standunsicherheiten im Sinne einer zerebellären Ataxie, einen rhythmischen Tremor und im weiteren Verlauf unnatürliches Lachen, weswegen die Krankheit auch Lachkrankheit genannt wird.
Entdeckung
Nachdem das Hochland von Papua-Neuguinea erst in den 1930er-Jahren überhaupt Kontakte mit der westlichen Zivilisation hatte, wurde die Krankheit in der zweiten Hälfte der 1950er Jahre erstmals beschrieben und untersucht. Besonders verdient machte sich dabei D. C. Gajdusek, der unter anderem bei Experimenten eine Übertragung von Kuru auf Affen nachweisen konnte, wofür ihm 1976 der Nobelpreis für Medizin verliehen wurde. Für die Krankheit, die damals unter den gut 10.000 Fore jährlich über 200 Opfer forderte, nahm man zunächst eine genetische Ursache an. Nachdem die genetische Hypothese aus epidemiologischen Gründen immer unwahrscheinlicher geworden war, blieb eine intensive Suche nach Umweltgiften oder Infektionsquellen ebenfalls erfolglos. Erst nachdem William J. Hadlow (1921–2015) die (neuropathologische) Ähnlichkeit mit der damals schon als übertragbar bekannten Scrapie erkannt hatte, untersuchte man die Übertragbarkeit der Kuru auf Affen unter langer Beobachtungszeit und war damit in den 1960er Jahren erfolgreich. Nach weiteren jahrzehntelangen medizinischen, epidemiologischen und anthropologischen Forschungen etablierte sich die Hypothese, dass Kuru bei den Fore durch Endokannibalismus (Verzehr von Fleisch verstorbener Stammesgenossen) und den im Zusammenhang damit stehenden Umgang mit hoch infektiösem Gehirn übertragen wurde.[1] Da der Kannibalismus 1954 aus anderen, nichtmedizinischen Gründen verboten worden war, nahm auch die Häufigkeit der Erkrankungen stetig ab, bis die Erkrankung gegen Ende des Jahrhunderts völlig verschwand.
Rückblickend wurde der Beginn der Epidemie um die Wende vom 19. zum 20. Jahrhundert zeitlich lokalisiert und ging vermutlich von einem einzigen (sporadischen) Fall aus. Frauen und Kinder, die sich beim Umgang mit dem infektiösen Gehirn auf parenteralem Weg infizierten, erkrankten vermutlich nach kurzer Inkubationszeit, während die alleinige perorale Aufnahme erst nach Jahrzehnten zu einer Erkrankung führte. Männer waren vermutlich allgemein weniger betroffen, da sie hauptsächlich Muskelfleisch zu sich nahmen.
Medizinische Bedeutung
Die Kuru ist medizinhistorisch von großem Interesse, wurde aber von einer breiteren Öffentlichkeit vor allem nach dem Auftreten von BSE und CJD in den 1990er-Jahren beachtet. 2009 entdeckten Mediziner, dass die Fore innerhalb kürzester Zeit eine genetische Mutation ausbildeten, die den Ausbruch der Krankheit verhindert. Zudem stellte sich später heraus, dass diese Mutation auch gegen alle anderen Transmissiblen Spongiformen Enzephalopathien (TSE) schützt. Die Forscher hoffen daraus Erkenntnisse für die Behandlung von weiteren degenerativen Hirnerkrankungen wie der Parkinson-Krankheit und der Alzheimer-Krankheit zu gewinnen.[2][3][4]
Meldepflicht
Transmissible spongiforme Enzephalopathien sind in Österreich gemäß § 1 Abs. 1 Nummer 1 Epidemiegesetz 1950 bei Verdacht, Erkrankung und Tod anzeigepflichtig. In Deutschland ist humane spongiforme Enzephalopathie (außer familiär-hereditärer Formen) gemäß § 6 Infektionsschutzgesetz (IfSG) bei Verdacht, Erkrankung und Tod seitens des Arztes usw. namentlich meldepflichtig.
Literatur
- N. Bons, N. Mestre-Frances, P. Belli, F. Cathala, D. C. Gajdusek, P. Brown: Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. In: Proc Natl Acad Sci U S A. 1999 Mar 30;96(7), S. 4046–4051.
- C. J. Gibbs Jr, D. M. Asher, A. Kobrine, H. L. Amyx, M. P. Sulima, D. C. Gajdusek: Transmission of Creutzfeldt-Jakob disease to a chimpanzee by electrodes contaminated during neurosurgery. In: Neurol Neurosurg Psychiatry. 1994 Jun;57(6), S. 757–758.
- J. Tateishi, P. Brown, T. Kitamoto, Z. M. Hoque, R. Roos, R. Wollman, L. Cervenakova, D. C. Gajdusek: First experimental transmission of fatal familial insomnia. In: Nature. 1995 Aug 3;376(6539), S. 434–435.
- J. Hinkelbein u. a.: Creutzfeldt-Jakob-Krankheit. In: Frank Wappler, Gerd Bürkle, Peter Tonner: Anästhesie und Begleiterkrankungen. Thieme-Verlag, Stuttgart/ New York 2006, ISBN 3-13-129941-X.
Einzelnachweise
- Walter Bruchhausen: Von der Bakteriologie zur molekularen Virologie und Prionenforschung. Die Entwicklung der Infektionslehre. In: Dominik Gross, H. J. Winkelmann (Hrsg.): Medizin im 20. Jahrhundert. Fortschritte und Grenzen. Ärztliche Praxis, München 2008, S. 6–25.
- Simon Mead u. a.: A Novel Protective Prion Protein Variant that Colocalizes with Kuru Exposure. In: The New England Journal of Medicine. Band 361 (2009), Nr. 21, S. 2056–2065.
- A naturally occuring variant of the human prion protein completely prevents prion disease
- Former brain-eating Papua tribe offers clues on deadly diseases