Creutzfeldt-Jakob-Krankheit
Die Creutzfeldt-Jakob-Krankheit (CJK) (englisch Creutzfeldt Jakob Disease, CJD) ist eine beim Menschen sehr selten auftretende, tödlich verlaufende und durch atypische Eiweiße (sogenannte Prionen) gekennzeichnete übertragbare spongiforme (mit schwammartiger Auflösung des Hirngewebes einhergehende) Enzephalopathie. Diese neurodegenerative Erkrankung kommt beim Menschen als übertragene, genetische oder sporadische Form vor. Charakteristisch für die Krankheit ist, dass die abnorm gefalteten Prionproteine vor allem im Gehirn den dort normalerweise vorhandenen „Vettern“ mit gesunder Struktur ihre veränderte Struktur aufzwingen und so dort einen verhängnisvollen biochemischen Prozess auslösen, der letztlich zu einer Degeneration des Gehirns führt. Die krankhaft gefalteten Proteine lagern sich in Nervenzellen ab und bilden Klumpen. Die Funktion der Nervenzellen wird zunehmend gestört, sodass es bis hin zum programmierten Zelltod kommt (Apoptose). Bei fortschreitender Erkrankung nimmt das befallene Gehirn eine schwammartig durchlöcherte Struktur mit fadenförmigen, proteinhaltigen Ablagerungen an. Im Blut eines erkrankten Menschen sind jedoch nur kleinste Mengen der infektiösen Prionen vorhanden.
| Klassifikation nach ICD-10 | |
|---|---|
| A81.- | Atypische Virus-Infektionen des Zentralnervensystems |
| A81.0 | Creutzfeldt-Jakob-Krankheit |
| F02.1* | Demenz bei Creutzfeldt-Jakob-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Die Benennung der Krankheit erfolgte zu Beginn der 1920er Jahre nach ihren Erstbeschreibern, den Nervenärzten Hans Gerhard Creutzfeldt (in Breslau) und (unabhängig davon) Alfons Maria Jakob (in Hamburg).
Für das Jahr 2017 meldete das Robert Koch-Institut für Deutschland 77 Erkrankungen, 2018 78 Fälle.[1]
Erstbeschreibung
Eine schwammartige (spongiforme) Auflösung von Gehirngewebe bei an Scrapie erkrankten Schafen war im Jahr 1732 in Großbritannien dokumentiert worden.[2]
Während eines Weiterbildungsaufenthaltes behandelte der spätere Kieler Neurologe Hans Gerhard Creutzfeldt 1913 an der Breslauer Universitätsnervenklinik unter Leitung von Alois Alzheimer eine junge Frau mit Sprachstörungen, Verwirrtheit und Muskelzuckungen, die kurze Zeit darauf verstarb. Durch den Ersten Weltkrieg konnte er diese „eigenartige, herdförmige Erkrankung des Zentralnervensystems“ erst 1920 publizieren[3], kurz vor dem Hamburger Neurologen Alfons Maria Jakob.[4]
Das Eponym geht auf den deutschen Neuropathologen Walther Spielmeyer zurück, der die Bezeichnung Creutzfeldt-Jakob-Krankheit 1922 vorschlug.[5]
CJK/CJD
Diese Erkrankung ist die häufigste beim Menschen vorkommende transmissible spongiforme Enzephalopathie (TSE). Die klassische CJK wird in drei bisher bekannte Formen unterteilt:
Sporadische Prionerkrankung (sCJK)
Die sporadische Form der Creutzfeldt-Jakob-Krankheit ist die häufigste weltweit beim Menschen auftretende Erkrankungsform. Die auslösenden Faktoren sind wahrscheinlich Prionen.
Häufigkeit
Die Erkrankung kommt weltweit mit einer ähnlichen Häufigkeit von etwa 1 Fall pro Jahr pro Million Einwohner vor, wird anfänglich jedoch oft fehldiagnostiziert, am häufigsten als virale Enzephalitis, paraneoplastische Kleinhirnatrophie, Depression, Vertigo oder Alzheimer-Krankheit.[6] In Deutschland beträgt das Geschlechtsverhältnis von Frauen zu Männern 2:1. Das Erkrankungsrisiko nimmt mit steigendem Alter zu, und der Erkrankungsgipfel liegt um das 70. Lebensjahr. In diesem Alter beträgt die jährliche Erkrankungswahrscheinlichkeit etwa 1:125.000. Danach sinkt das Risiko wieder ab. Vereinzelt erkranken auch jüngere Menschen. In Deutschland erkranken jährlich etwa 7 Menschen, die jünger als 50 Jahre alt sind, an einer sporadischen CJK. Es können selbst Jugendliche erkranken, obgleich weltweit bisher nur eine Handvoll derartiger Fälle beschrieben wurde. Die Wahrscheinlichkeit, bereits vor dem 30. Lebensjahr zu erkranken, beträgt etwa 1:3.000.000.
Krankheitsverlauf/Symptome
Die Erkrankung beginnt zunächst schleichend, doch ein Erkrankter verliert unaufhaltsam und rasch fortschreitend seine geistigen und motorischen Fähigkeiten. Folgende Symptome können beobachtet werden: Schreckhaftigkeit, motorische Störungen (Myoklonien, Ataxie), Gedächtnisstörungen, Störungen der Wahrnehmung (Halluzinationen) und der Vigilanz, visuelle Störungen und Persönlichkeitsveränderungen, vegetative Störungen und Verwirrtheit bis hin zur Demenz. Das Spätstadium der Erkrankung ist durch den akinetischen Mutismus gekennzeichnet. In der Regel führt die Erkrankung innerhalb weniger Monate zum Tod. Die Erkrankungsdauer kann von 3–6 Wochen bis zu mehr als 2 Jahren betragen. Die mittlere Erkrankungsdauer beträgt 4–6 Monate.
Bei der Brownell-Oppenheimer-Form, die auch als cerebelläre (Kleinhirn-)Variante bezeichnet wird, kommt es im ersten Monat der Erkrankung nur zu Zeichen einer Kleinhirnstörung ohne kognitive Einschränkungen. Etwa 20 % der sporadischen Erkrankungen gehören zu dieser Form. Bei einem mittleren Erkrankungsalter von 63 Jahren treten zunächst meist Gangunsicherheit, Schwindel und Koordinationsstörungen auf, aber nach im Mittel drei Monaten bildet sich ebenfalls die progrediente Vollform einschließlich kognitiver Störungen aus. Trotzdem sind auch bei der cerebellären Form in der Kernspintomographie in den Diffusions- und T2-gewichteten FLAIR-Sequenzen meist keine Hyperintensitäten im Kleinhirn zu finden, jedoch in den Basalganglien und im Thalamus.[7]
Genetische Prionerkrankung
In dieser Form wird eine ganze Gruppe von familiär vererbbaren Erkrankungen zusammengefasst. Bei all diesen Formen wird eine spezifische Mutation vererbt, welche zu einem fehlerhaften Prion-Protein führt. Diese Krankheitsgruppe ist sehr uneinheitlich (heterogen) und durch sehr variable klinische Symptome gekennzeichnet. Der Erkrankungsgipfel liegt hier insgesamt um das 50. Lebensjahr und damit früher als bei der sporadischen Form. Auch die Erkrankungsdauer ist häufig länger. Zu diesen Erkrankungsformen zählen die familiäre/genetische Creutzfeldt-Jakob-Krankheit, das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die tödliche familiäre Schlaflosigkeit (fatal familial insomnia, FFI). Die familiäre Form der Creutzfeldt-Jakob-Krankheit ist bereits 1930 von Friedrich Meggendorfer beschrieben worden.[8][9]
Übertragene Formen
Eine direkte Übertragung des Erregers von Mensch zu Mensch ist bisher nur auf iatrogenem Wege (durch Ärzte verursacht) über Kontakt mit infektiösem Gewebe nachgewiesen worden. Dies geschah besonders früher durch Hirnhaut- und Augenhornhauttransplantate sowie durch unzureichend sterilisierte neurochirurgische Instrumente. Außerdem wurde eine direkte Übertragung bei aus Leichenhypophysen extrahierten Wachstumshormonen bzw. Gonadotropinen beobachtet. Weltweit sind insgesamt 132 Fälle einer Infektion durch Wachstumshormonpräparate bekannt, wobei die meisten dieser Fälle aus Frankreich und Großbritannien berichtet wurden. In Deutschland wurde bislang noch kein Fall bekannt, obwohl auch dort kleinwüchsige Patienten mit Wachstumshormonen behandelt wurden.
Die meisten Fälle, die mit Hirnhauttransplantaten in Verbindung gebracht werden, traten in Japan auf. Diese Erkrankungen werden fast ausschließlich auf das deutsche Produkt Lyodura von der B. Braun Melsungen AG zurückgeführt. Aufgrund mangelnder Kontrollen der Hirnhautspender sowie des Herstellungsprozesses, bei dem Hirnhäute ungenügend desinfiziert und in Stapeln übereinandergelagert wurden, wodurch es zu einer Querkontamination gesunder Hirnhäute mit Prionen kam, galt dieses Produkt als besonders gefährlich. Lyodura wurde als eine Art „Pflaster“ nicht nur zur Rekonstruktion der Hirnhaut, sondern auch in einer Vielzahl nicht-neurochirurgischer Operationen verwendet, zumal es sich durch geringe Abstoßungsreaktionen auszeichnete. Lyodura musste 1996 aus dem Verkehr gezogen werden.
Neue Variante der Creutzfeldt-Jakob-Krankheit
In Großbritannien wurde am 20. März 1996 bekanntgegeben, dass mehrere junge Menschen an einer neuen Variante von CJD (nvCJD) gestorben waren.
Übertragung
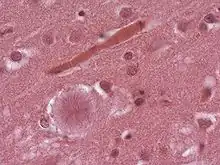
Nach aktuellen Erkenntnissen besteht eine Wahrscheinlichkeit von 99 % dafür, dass diese Variante (heute als nvCJD bekannt, nv = new variant = neue Variante) durch den Verzehr von BSE-verseuchtem Rindfleisch hervorgerufen wird. Vermutlich ist allerdings eine Mehrheit der Bevölkerung gegen die Ansteckung durch BSE-verseuchte Nahrung resistent, denn alle bisherigen nvCJD-Erkrankten hatten eine genetische Veranlagung, die sich nur bei knapp 40 Prozent der europäischen Bevölkerung findet.[10] An einer kritischen Stelle des Gens, welches das Prionen-Eiweiß kodiert, fand sich bei ihnen stets nur die Anweisung zum Einbau der Aminosäure Methionin. Der überwiegende Teil der Bevölkerung ist jedoch mischerbig und besitzt zusätzlich ein Gen, das den Einbau von Valin an dieser Stelle bewirkt. Es liegt die Schlussfolgerung nahe, dass sich menschliche Prionen leichter von BSE-Prionen umfalten lassen, wenn sie an der bezeichneten Stelle die Aminosäure Methionin enthalten.
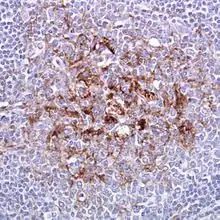
Im Unterschied zur sporadischen Erkrankung (sCJD), die vom Gehirn selbst ausgeht und im Wesentlichen auf das Zentralnervensystem beschränkt bleibt (geringe Mengen des abnormen Prionproteins wurden auch in Nerven und Muskeln nachgewiesen), bezieht die new variant CJD (nvCJD) auch das sogenannte lymphoretikuläre System mit Lymphknoten, Milz und Tonsillen (Mandeln) mit ein. Eine Tonsillenbiopsie, die chirurgische Probenentnahme aus den Gaumenmandeln, kann daher zur Diagnose der nvCJD verwendet werden. Die Tonsillen sind bei nvCJD immer positiv, während sie bei sporadischer CJD (sCJD), bei iatrogener CJD (Mensch zu Mensch-Übertragung durch Dura-Transplantate oder Hypophysenextrakt) und bei den erblichen Formen immer negativ sind.
Es gibt außerdem Hinweise darauf, dass Menschen grundsätzlich auch über Bluttransfusionen mit der neuen Variante der Creutzfeldt-Jakob-Krankheit (nvCJD) infiziert werden können. In Großbritannien gibt es einige wenige auffällige Einzelfälle, in denen es sehr plausibel erscheint, dass die Erkrankung auf diesem Wege übertragen worden ist. Auch Tierversuche deuten auf eine hohe Wahrscheinlichkeit dieser Möglichkeit hin. Mit 100%iger Sicherheit nachgewiesen werden konnte dieser Übertragungsweg allerdings bis jetzt dennoch nicht. Aus der Möglichkeit der Ansteckung über Bluttransfusionen (und damit also Blut bzw. Blutbestandteilen an sich) wird vermutet, dass es bei einer Verkettung unglücklicher Umstände und Zufällen in der Vergangenheit in Großbritannien in seltenen Einzelfällen zu Infektionen bei Operationen gekommen sein könnte. Als Ursache für diese unwahrscheinlichen Infektionsfälle wurden neben Bluttransfusion auch OP-Besteck vermutet. Begründet wird dies damit, dass in der Vergangenheit teilweise Desinfektionstechniken verwendet wurden, bei denen nicht mit Sicherheit ausgeschlossen werden kann, dass Prionen die Prozedur und anschließende Lagerung der Geräte überdauerten. Allerdings ist bisher kein einziger Fall bekannt, in dem ein konkreter Verdacht auf diesen Übertragungsweg vorlag, es handelt sich um eine reine Vermutung hinsichtlich eines denkbaren Übertragungsweges. Ob es jemals einen entsprechenden Fall geben wird, ist nicht vorauszusagen.
Neue Forschungsergebnisse an der Universität Zürich lassen den Schluss zu, dass eine Übertragung über Aerosole in der Luft möglich ist. Allerdings dürfte diese Ansteckungsform in der Praxis höchstens bei der Arbeit mit den Kadavern erkrankter Tiere in Schlachthöfen und Laboren eine Rolle spielen.[11]
Diagnose
Im August 2005 gaben der Neurologe Claudio Soto und seine Kollegen von der University of Texas (USA) bekannt, dass nunmehr die Rinderseuche BSE und die neue Variante der Creutzfeldt-Jakob-Krankheit mit einem Bluttest zu diagnostizieren ist. Die Eigenschaft der abnorm veränderten infektiösen Prionen, ihre Struktur anderen gesunden Prionen aufzuzwingen, nutzten die Forscher aus, um die im Blut von Erkrankten nur in verschwindend geringer Zahl vorhandenen infektiösen Prionen um den Faktor zehn Millionen zu vermehren und damit leicht nachweisbar zu machen. In Versuchsreihen mit Hamstern ließen sich so die infektiösen Prionen mit einer Zuverlässigkeit von 89 % und ohne Fehlalarm nachweisen. An der Anwendbarkeit auch für die Diagnose beim Menschen und einer Kontrolle von Blutspenden wird gearbeitet.
Das falsch gefaltete Protein ist gegen die Aufspaltung durch Proteasen resistent, nicht jedoch die normale Form von PrP. In der Diagnostik können also die normalen Proteine „verdaut“ werden; und wenn Reste auftreten, dann muss es sich um die pathogene Form des Proteins handeln.
Abgesehen davon lassen sich nvCJD-Prionen außerdem im Mandel-, Milz- oder Blinddarmgewebe nachweisen, lange bevor sie das Gehirn befallen. Zur Erhärtung des nvCJD-Verdachts kann daher eine Mandelbiopsie durchgeführt werden.
Magnetresonanztomographie
Bei der Diagnose ist die Magnetresonanztomographie die bildgebende Untersuchung der Wahl bei klinisch vermuteten nvCJD. In über 90 % der neuropathologisch bestätigten nvCJD-Fälle zeigte sich schon in frühen Stadien der Erkrankung (2 bis 10 Monate nach Beginn der Symptome) im Thalamus das sogenannte Pulvinarzeichen. Dies ermögliche in den meisten Fällen die Diagnose, ohne dass weitere Untersuchungen erforderlich seien.[12]
EEG
Die Elektroenzephalografie zeigt in den meisten Fällen diffuse Verlangsamungen, kann jedoch bis zum Auftreten von psychiatrischen oder neurologischen Symptomen unauffällig sein.
Liquor cerebrospinalis
Die Liquoruntersuchung ist in den Standardparametern unauffällig, es finden sich eine normale Zellzahl, Gesamteiweiß und Glukose, nur selten eine leichte bis mittelgradige Schrankenstörung. Der Nachweis des Proteins 14-3-3 im Liquor hat sich als zuverlässiger und sensitiver Marker für die sporadische Creutzfeldt-Jakob-Krankheit (sCJK) erwiesen. Bei nvCJK ist dieser jedoch nur in 50 % positiv. Es wurde jedoch festgestellt, dass eine erhöhte Konzentration von CSF-Tau ein empfindlicher Marker für nvCJD ist. Ein negatives 14-3-3 hat einen negativen Vorhersagewert von 63 % und ein negatives Tau hat einen negativen Vorhersagewert von 81 %. Wenn beide Tests negativ sind, steigt der negative Vorhersagewert für vCJD auf 84 %[13]
Krankheitsverlauf und Symptome
Das mittlere Alter zu Beginn beträgt 26 Jahre (Bereich 12 – 74 Jahre). Die Krankheitsdauer ist mit 14 Monaten (6–39) ungewöhnlich länger als bei der sporadischen Form. Die meisten Patienten wurden im Verlauf der Krankheit zuerst von einem Psychiater untersucht. Die frühen Stadien der Creutzfeldt-Jakob-Variante werden von psychiatrischen Symptomen dominiert, aber neurologische Symptome gehen in 15 % der Fälle psychiatrischen Symptomen voraus und treten in 22 % der Fälle ab Krankheitsbeginn in Kombination mit psychiatrischen Symptomen auf. Häufige frühpsychiatrische Merkmale waren Dysphorie, Anergie, Interessensverlust, Schlaflosigkeit, Angstzustände und Entzug, was in vielen Fällen zur Diagnose einer Depression führte, aber eine Minderheit der Fälle entwickelte auch psychotische Merkmale wie auditive oder visuelle Halluzinationen und Paranoidität oder Wahnvorstellungen. Viele Patienten wurden aufgeregt oder aggressiv, was manchmal zu Schwierigkeiten beim Management führte, aber Suizidgedanken waren selten (9 % der Fälle), und es gab in keinem Fall Aufzeichnungen über absichtliche Selbstverletzungen. Flüchtige Wahnvorstellungen wurden in früheren Berichten als ungewöhnliches psychiatrisches Merkmal beschrieben und obwohl in einigen Fällen eindeutig ein Merkmal, waren sie im Vergleich zur Häufigkeit anderer offensichtlicherer psychiatrischer Symptome relativ selten.[14][15] Die Möglichkeit einer zugrunde liegenden neurologischen Störung wurde in vielen Fällen durch die Entwicklung einer kognitiven Beeinträchtigung, einschließlich schlechtem Gedächtnis, Konzentrationsstörungen, Orientierungslosigkeit oder in einer Minderheit offener Verwirrung, erhöht. Diese Merkmale entwickelten einen Median von 4 bis 7,5 Monaten ab klinischem Beginn, obwohl sie in einer kleinen Minderheit der Fälle bereits in den frühesten Stadien vorhanden waren. Das häufigste frühe neurologische Merkmal waren Schmerzen, die nicht mit sensorischen Symptomen verbunden waren. Dies war hartnäckig und unangenehm und betraf häufig die Gliedmaßen, den Rumpf oder das Gesicht. Zwischen einem Median von vier und sechs Monaten umfassten häufige neurologische Merkmale Gangstörungen, häufig in Form einer geringfügigen Unstetigkeit, und Dysarthrie. Parästhesien und Taubheitsgefühle in ähnlicher Verteilung wie Schmerzen traten häufig im Median von 4 bis 6 Monaten auf und betrafen fast die Hälfte der Patienten.[16] Die Kombination einer psychiatrischen Störung mit affektiven oder psychotischen Merkmalen und anhaltenden Schmerzen, Dysarthrie, Gangataxie oder sensorischen Symptomen sollte zumindest den Verdacht auf eine Variante der Creutzfeldt-Jakob-Krankheit erwecken, insbesondere wenn dies mit einem Hinweis auf eine kognitive Beeinträchtigung verbunden ist. Einige der neurologischen Merkmale, wie sensorische Symptome, Gangschwankungen und Dysarthrie, können bei psychiatrischen Erkrankungen oder als Nebenwirkungen von Psychopharmaka auftreten, aber das Fortbestehen dieser Symptome und die Entwicklung zusätzlicher neurologischer Symptome können auf die Variante Creutzfeldt-Jakob hinweisen.
Neurologische Merkmale der Creutzfeldt-Jakob-Variante wie Kleinhirnzeichen, unwillkürliche Bewegungen (Myoklonus, Chorea oder Dystonie), Zeichen des oberen Motoneurons und visuelle Symptome sind sehr häufig, treten jedoch relativ spät im Verlauf der Krankheit auf. Psychiatrische Symptome, die auf die Wahrscheinlichkeit einer organischen Ätiologie hinweisen, z. B. Orientierungslosigkeit, Halluzinationen und beeinträchtigte Selbstversorgung, treten ebenfalls spät auf.
Im Endstadium haben die Patienten der Krankheit keinerlei Möglichkeit mehr, Kontakt mit ihrer Umwelt aufzunehmen oder auf einen solchen zu reagieren. Darum werden nvCJD-Kranke im Endstadium der Krankheit oft als „The Living Dead“ (Die lebenden Toten) bezeichnet. Manchmal tritt hierbei eine vollständige spastische Lähmung des Körpers, die sogenannte Enthirnungsstarre, ein. Die Patienten verweilen recht lange in diesem Endzustand der Erkrankung (terminalen Zustand), bis sie insbesondere an einer Lungenentzündung oder durch Atemlähmung sterben.
Häufigkeit
Bis zum Februar 2015 waren in Großbritannien 177 Menschen an nvCJD verstorben sowie außerhalb Großbritanniens weitere 52 Personen, die Hälfte davon in Frankreich.[17][18] Die Zahl der Erkrankungen nimmt ab, daher gilt die Gefahr als gebannt. Noch 2005 wurde vermutet, dass bis 2015 eine massive Epidemie des „menschlichen Rinderwahnsinns“ Großbritannien heimsuchen wird und womöglich viele Tausend Menschen dieser Krankheit erliegen werden. Erhärtet wurde diese Vermutung auch durch eine Studie, bei der durch Untersuchungen von entferntem Mandel- und Blinddarmgewebe festgestellt wurde, dass mehrere Tausend Briten den nvCJD-Erreger in sich tragen müssen.
Die BBC berichtete am 12. Januar 2005 von Ergebnissen einer Gruppe von Wissenschaftlern, nach denen eine große Epidemie unwahrscheinlich ist. Dies wird u. a. dadurch gestützt, dass die Zahl der Todesfälle in Großbritannien von 28 im Jahr 2000 auf neun im Jahr 2004 zurückgegangen ist.
Meldepflicht
In der Schweiz ist der klinische Verdacht auf eine Form von Creutzfeldt-Jakob-Krankheit (CJK), der Tod eines Patienten und die Bestätigung der Erkrankung durch Autopsie meldepflichtig und zwar nach dem Epidemiengesetz (EpG) in Verbindung mit der Epidemienverordnung und Anhang 1 bzw. Anhang 2 der Verordnung des EDI über die Meldung von Beobachtungen übertragbarer Krankheiten des Menschen.
Transmissible spongiforme Enzephalopathien sind in Österreich gemäß § 1 Abs. 1 Nummer 1 Epidemiegesetz 1950 bei Verdacht, Erkrankung und Tod anzeigepflichtig. Zur Anzeige verpflichtet sind unter anderen Ärzte und Labore (§ 3 Epidemiegesetz).
In Deutschland ist humane spongiforme Enzephalopathie (außer familiär-hereditärer Formen) gemäß § 6 Infektionsschutzgesetz (IfSG) bei Verdacht, Erkrankung und Tod seitens des Arztes usw. namentlich meldepflichtig. Der Kreis der Meldepflichtigen richtet sich nach § 8 IfSG, was zu melden ist nach § 9 IfSG.
Literatur
- S1-Leitlinie Creutzfeldt-Jakob-Krankheit der Deutschen Gesellschaft für Neurologie. In: AWMF online (Stand 2012)
Weblinks
- Creutzfeldt-Jakob-Krankheit / Variante Creutzfeldt-Jakob-Krankheit – Informationen des Robert Koch-Instituts
- Prionforschungsgruppe Göttingen (Deutsche CJD-Surveillance)
- Prognose zur vCJD Ausbreitung aus dem Jahr 2001 – Focus-Interview mit Hans A. Kretzschmar
- Eichhörnchen-Hirn als mögliche Infektionsquelle
- BBC-Bericht (englisch) über die Wahrscheinlichkeit einer größeren Epidemie
- Voraussage der Entwicklung der nvCJD-Fallzahlen in Frankreich (derzeit -wann?- das am stärksten betroffene Land)
- CJK-Initiative e.V. – Eine Initiative von Angehörigen für Angehörige (gemeinnütziger Verein).
- CJD Support Network – A patient support group (UK).
Einzelnachweise
- Robert Koch-Institut, Robert Koch-Institut: Infektionsepidemiologische Jahrbuch meldepflichtiger Krankheiten für 2018. Robert Koch-Institut, 26. Juli 2019, S. 67 ff., doi:10.25646/5978 (rki.de [abgerufen am 20. August 2020]).
- Sabine Schuchart: Creutzfeldt und Jakob waren beide einem Rätsel auf der Spur. (= Berühmte Entdecker von Krankheiten. 60) In: Deutsches Ärzteblatt. Band 116, Heft 49, 6. Dezember 2019 (Schlusspunkt).
- H. G. Creutzfeldt: Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Vorläufige Mitteilung. In: Arch Psychiatrie Nervenkrankh. Band 1920, 57, S. 1–18.
- A. Jakob: Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswerten anatomischen Befunde (spastische Pseudosklerose-Encephalomyelopathie mit disseminierten Degenerationsherden). Vorläufige Mitteilung. In: Dtsch Z Nervenheilk. Band 70, 1921, S. 132–146.
- Sabine Schuchert: Creutzfeldt und Jakob waren beide einem Rätsel auf der Spur. In: Deutsches Ärzteblatt. Jahrgang 116, 2019, Heft 49 vom 6. Dezember, S. 60. Link abgerufen am 15. Dezember 2019, 21:18 Uhr CEST
- Ross W. Paterson, Charles C. Torres-Chae, Amy L. Kuo, ...: Differential Diagnosis of Jakob-Creutzfeldt Disease. In: Archives of neurology. Band 69, Nr. 12, 21. Januar 2017, S. 1578–1582, doi:10.1001/2013.jamaneurol.79, PMC 4401069 (freier Volltext).
- Bart K. Chwalisz, Bradley R. Buchbinder, Jeremy D. Schmahmann, Wesley R. Samore: Case 32-2019: A 70-Year-Old Woman with Rapidly Progressive Ataxia New England Journal of Medicine 2019, Band 381, Ausgabe 16 vom 17. Oktober 2019, Seiten 1569–1578, DOI: 10.1056/NEJMcpc1909624
- Friedrich Meggendorfer: Klinische und genealogische Beobachtungen bei einem Fall von spastischer Pseudokosklerose Jakobs. In: Z Neurol Psychiatry. Band 128, 1930, S. 337–341. doi:10.1007/BF02864269
- P. Gambetti, Q. Kong, W. Zou, P. Parchi, S. G. Chen: Sporadic and familial CJD: classification and characterisation. In: Br Med Bull. Band 66, 2003, S. 213–239. doi:10.1093/bmb/66.1.213. PMID 14522861.
- Tzehow M et al. (2017): Variant Creutzfeldt-Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. Correspondence. N Engl J Med 2017; 376:292-294 January 19, 2017, doi:10.1056/NEJMc1610003
- Johannes Haybaeck, Mathias Heikenwalder u. a.: Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. In: PLoS Pathogens. 7, 2011, S. e1001257, doi:10.1371/journal.ppat.1001257.
- Donald A. Collie, David M. Summers, Robin J. Sellar, James W. Ironside, Sarah Cooper: Diagnosing Variant Creutzfeldt-Jakob Disease with the Pulvinar Sign: MR Imaging Findings in 86 Neuropathologically Confirmed Cases. In: American Journal of Neuroradiology. Band 24, Nr. 8, 1. September 2003, ISSN 0195-6108, S. 1560–1569, PMID 13679271 (ajnr.org [abgerufen am 13. August 2020]).
- A. J. Green, E. J. Thompson, G. E. Stewart, M. Zeidler, J. M. McKenzie: Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. In: Journal of Neurology, Neurosurgery, and Psychiatry. Band 70, Nr. 6, Juni 2001, ISSN 0022-3050, S. 744–748, doi:10.1136/jnnp.70.6.744, PMID 11385008, PMC 1737395 (freier Volltext) – (nih.gov [abgerufen am 13. August 2020]).
- M. Zeidler, E. C. Johnstone, R. W. Bamber, C. M. Dickens, C. J. Fisher: New variant Creutzfeldt-Jakob disease: psychiatric features. In: Lancet (London, England). Band 350, Nr. 9082, 27. September 1997, ISSN 0140-6736, S. 908–910, doi:10.1016/s0140-6736(97)03148-6, PMID 9314868 (nih.gov [abgerufen am 14. August 2020]).
- R. G. Will, G. Stewart, M. Zeidler, M. A. Macleod, R. S. G. Knight: Psychiatric features of new variant Creutzfeldt-Jakob disease. In: Psychiatric Bulletin. Band 23, Nr. 5, Mai 1999, ISSN 0955-6036, S. 264–267, doi:10.1192/pb.23.5.264 (cambridge.org [abgerufen am 14. August 2020]).
- M.-A. Macleod, G. E. Stewart, M. Zeidler, R. Will, R. Knight: Sensory features of variant Creutzfeldt-Jakob disease. In: Journal of Neurology. Band 249, Nr. 6, 1. Juni 2002, ISSN 1432-1459, S. 706–711, doi:10.1007/s00415-002-0696-2.
- Andy Coghlan: When the cows went mad. In: New Scientist. Band 225, Nr. 3010, 2015, S. 48.
- onmeda.de: Variante der Creutzfeldt-Jakob-Krankheit (vCJK): Definition, Stand: 17. Dezember 2014.