Refsum-Syndrom
Die Refsum-Kahlke-Krankheit (Synonyme: Refsum-Syndrom, Refsum-Thiébaut-Krankheit, Heredopathia atactica polyneuritiformis, Morbus Refsum) ist ein Gendefekt, durch den der Körper nicht fähig ist Phytansäure adäquat abzubauen, sodass sich Phytansäure im Körper anreichert. Diese peroxisomale Stoffwechselstörung wird autosomal-rezessiv vererbt und nach dem Erstbeschreiber, dem norwegischen Arzt Sigvald Refsum (1907–1991) benannt. Diese Form der Heredoataxie wird zu den hereditären motorisch-sensiblen Neuropathien gerechnet (Typ IV).
| Klassifikation nach ICD-10 | |
|---|---|
| G60.1 | Refsum-Kahlke-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Ursache, Symptome und Verlauf
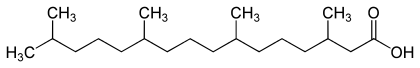
1963 gelang dem heute in Hamburg lehrenden Professor Winfried Kahlke der erste biochemische Nachweis einer hereditären Fettstoffwechselstörung, als er das Substrat des Refsum-Syndroms, die Phytansäure, identifizierte.[1][2] Kahlke fand mittels Gaschromatografie die exogene Ursache der erblichen Lipidstoffwechselstörung, ein mit der Nahrung aufgenommenes Abbauprodukt des Chlorophylls, die 3.7.11.15-Tetramethyl-hexadecansäure (Phytansäure). Diese Entdeckung beflügelte die Stoffwechselforschung weltweit.[3]
Die hauptsächliche biochemische Abnormalität ist die Akkumulation von Phytansäure, einer gesättigten, verzweigtkettigen Fettsäure, die ausschließlich mit der Nahrung aufgenommen wird. Da sich bei Phytansäure an der β-Position eine Methylgruppe befindet, kann diese Fettsäure nicht direkt durch mitochondriale oder peroxisomale β-Oxidation metabolisiert werden. Demnach wird sie der peroxisomalen α-Oxidation unterzogen, für die das Enzym Phytanoyl–CoA-Hydroxylase verantwortlich ist. Ist dieses Enzym defekt, kommt es zum klinischen Erscheinungsbild des Morbus Refsum. Die Krankheit ist genetisch heterogen: Das defekte Enzym ist entweder die Phytanoyl–CoA-Hydroxylase selbst oder aber Peroxin-7, das für den Transport der Phytanol–CoA-Hydroxylase in die Peroxisomen verantwortliche Transportprotein.
Die Hauptmerkmale des Syndroms sind Retinopathia pigmentosa mit Nachtblindheit und progredienten Gesichtsfeldeinschränkungen, eine periphere Polyneuropathie, eine zerebelläre Ataxie und eine sich entwickelnde Taubheit. Andere häufigere Symptome sind Nystagmus, Ichthyose, Katarakte, ein Verlust des Geruchssinns (Anosmie) sowie Skelettdeformitäten bei Dysplasie der Epiphysen.
Erstes Symptom ist häufig die Nachtblindheit. Der Verlauf ist schubförmig mit Teilremissionen und akuten Exazerbationen vor allem bei metabolischem Stress während Operationen, Infekten, verminderter Zufuhr von Nahrungsenergie, Schwangerschaft oder schweren Erkrankungen, bei denen viel Phytansäure aus Leber und Körperfettgewebe freigesetzt wird. Die Symptomatik beginnt in einem Alter von der frühen Kindheit bis zu 50 Jahren, meistens aber im 2. oder 3. Lebensjahrzehnt. Männer und Frauen sind zu gleichen Teilen betroffen. Unbehandelt kommt es zu Ertaubung und Erblindung etwa um das 40. Lebensjahr. Lebensgefährlich können die während der Exazerbationen auftretenden akuten Herz-Rhythmusstörungen sein.
Infantile Form
Neben der Adulten Form gibt es auch eine Infantile Form (IRD), die klinisch mehr dem Zellweger-Syndrom ähnelt. Sie gilt als mildeste Ausprägung des Peroxisomenbiogenesedefekte-Zellweger-Syndrom-Spektrums (PBD-ZSS).[4]
Diagnosestellung
Die Diagnose wird durch den laborchemischen Nachweis der Phytansäure in Plasma und Urin gestellt. Auch gesunde Menschen, die heterozygote Anlageträger sind, können dadurch erkannt werden.
Differentialdiagnose
Abzugrenzen ist unter anderem das Flynn-Aird-Syndrom.
Behandlung
Ein Heilverfahren für das Refsum-Syndrom ist noch nicht bekannt.[5] Die Erkrankung kann aber durch eine konsequente phytansäurearme Diät weitestgehend zum Stillstand gebracht werden. Hierbei wird empfohlen, die aufgenommene Phytansäuremenge unter 10 mg/Tag zu halten (die Menge der mit einer normalen Ernährung aufgenommenen Phytansäure beträgt etwa 100 mg/Tag). Nahrungsmittel mit einem hohen Phytansäuregehalt sind insbesondere Fleisch von Wiederkäuern wie Rindern und Molkereiprodukte. Neben einer vitaminreichen phytansäurearmen Ernährung ist auch eine ausreichende Kalorienzufuhr wichtig, um eine unkontrollierte Mobilisierung von Phytansäure aus dem Fettgewebe zu vermeiden.
Bei Einhalten der Diät kann die periphere Neuropathie zum Stillstand kommen und auch die Hautveränderungen können abheilen; im Bereich von Auge und Ohr schreitet die Erkrankung zumindest nicht weiter fort. Wenn diätetische Maßnahmen für eine zufriedenstellende Senkung der Konzentration der Phytansäure im Blutplasma nicht ausreichen, kann dies durch regelmäßige Plasmapherese oder die selektive Lipidapherese erreicht werden.[6][7][8] Bei der Lipidapherese macht man sich dabei zu Nutze, dass Phythansäure im Blutplasma an Cholesterin gebunden und mit diesem zusammen entfernt wird.[9]
Einzelnachweise
- E. Klenk, W. Kahlke: Über das Vorkommen der 3.7.11.15-Tetramethyl-hexadecansäure (Phytansäure) in den Cholesterinestern und anderen Lipoidfraktionen der Organe bei einem Krankheitsfall unbekannter Genese (Verdacht auf Heredopathia atactica polyneuritiformis [Refsum-Syndrom]). In: Hoppe Seylers Z Physiol Chem. 333, 1963, S. 133–139.
- W. Kahlke: Refsum-Syndrom. Lipoidchemische Untersuchungen bei 9 Fällen. In: Klin Wochenschr. 42, 1964, S. 1011–1016.
- Karl F. Masuhr: Die Refsum-Kahlke-Krankheit. In: Akt Neurol. 40 (01), 2013, S. 58.
- Refsum-Krankheit, infantile Form. In: Orphanet (Datenbank für seltene Krankheiten).
- Thomas M. Devlin (Hrsg.): Textbook of Biochemistry with Clinical Correlations. 6. Auflage. Wiley & Sons, 2006, ISBN 0-471-67808-2, S. 686.
- J. P. Hungerbühler u. a.: Refsum's disease: management by diet and plasmapheresis. In: Eur Neurol. 24(3), 1985, S. 153–159. PMID 2581787.
- F. B. Gibberd u. a.: Heredopathia atactica polyneuritiformis (refsum's disease) treated by diet and plasma-exchange. In: The Lancet. 1(8116), 1979, S. 575–578. PMID 85164.
- K. Rüther: Adult Refsum disease. A retinal dystrophy with therapeutic options. In: Ophthalmologe. 102(8), 2005, S. 772–777. PMID 15990985.
- D. Zolotov, S. Wagner, K. Kalb, J. Bunia, A. Heibges, R. Klingel: Long-term strategies for the treatment of Refsum's disease using therapeutic apheresis. In: J Clin Apher. 27(2), 2012, S. 99–105. PMID 22267052 .
Weblinks
- Krankheitsbild bei der Selbsthilfevereinigung Pro Retina Deutschland
- Refsum-Syndrom. In: Online Mendelian Inheritance in Man. (englisch).
- Refsum-Syndrom in Orphanet