Adrenoleukodystrophie
Adrenoleukodystrophie (X-ALD) oder Addison-Schilder-Syndrom ist eine vererbliche Stoffwechselkrankheit, die meist im Kindesalter auftritt und oft einen schnellen neurologischen Verfall mit sich bringt. Im Endstadium zeigt sich eine ausgeprägte Demenz, die schließlich zum Verlust der lebenswichtigen Körperfunktionen und damit zum Tode führt. Da die Erkrankung x-chromosomal-rezessiv vererbt wird, sind fast ausschließlich Männer betroffen. Frauen sind meist nur Überträgerinnen (Konduktor) der Genmutation. Es kommt nur sehr selten vor, dass beide X-Chromosomen einer Frau den Defekt tragen, was dann zur Ausprägung der Krankheitssymptome führt.
| Klassifikation nach ICD-10 | |
|---|---|
| E71.3 | Störungen des Fettsäurestoffwechsels Adrenoleukodystrophie Addison-Schilder-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Die Prävalenz liegt beim männlichen Geschlecht bei etwa 1:20.000. Die Adrenoleukodystrophie ist weltweit in allen Bevölkerungskreisen in etwa gleich häufig.[1]
Ursachen
Genetische Untersuchungen haben ergeben, dass alle Betroffenen Mutationen in dem ABCD1-Gen aufweisen, das ein Gen aus der Gruppe der ABC-Transporter ist. Dieses Gen befindet sich auf dem X-Chromosom, Genlocus q28.[1] Es kodiert das peroxisomale ABC-Halbtransporter-ALD-Protein, ein Protein, welches in der Membran der Peroxisomen lokalisiert ist. Es ist umstritten, ob es direkt am Transport von überlangkettigen Fettsäuren in die Peroxisomen beteiligt ist. Diese Fette werden normalerweise innerhalb des Peroxisoms abgebaut. Bei X-ALD-Patienten kommt es jedoch zu einer Anhäufung der Fettsäuren vor allem in der Nebennierenrinde und in der weißen Gehirnsubstanz (daher von lat.: adreno ‚die Nebenniere betreffend‘; von griech. leukos ‚weiß‘, dys ,falsch‘ und trophein ‚ernähren‘ mit -dystrophie im Sinne von ‚Funktionsstörung‘), sowie in den Leydig-Zellen im Hoden. Überlangkettige Fettsäuren werden auch in Zellmembranen eingelagert. Daher vertreten manche Wissenschaftler die Hypothese, dass es bei X-ALD-Patienten zu einer Veränderung der Membranstruktur des Myelins kommt. Dies könnte eine Ursache der Demyelinisierung darstellen und letztlich die Weiterleitung von Impulsen behindern und den geistigen und motorischen Verfall der Patienten bedingen.
Diagnostik
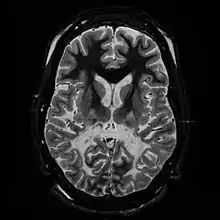
Klinisch ist die Adrenoleukodystrophie heterogen, mit verschiedenen klar unterscheidbaren Formen (Phänotypen), die aber keine klare Korrelation zum Genotyp haben. Mit etwa 35 % ist bei Jungen unter 12 Jahren der zerebral-entzündliche Typ, auch Zerebrale Adrenoleukodystrophie genannt, die häufigste Form.[2] Diese führt innerhalb von höchstens zwei Jahren nach Diagnosestellung zu schwerster Behinderung oder Tod.
Labordiagnostisch kann eine Erhöhung der überlangkettigen Fettsäuren, v. a. von Cerotinsäure (C26:0) und den Quotienten C26:0/C24:0, C26:0/C22:0 und C24:0/C22:0 im Serum festgestellt werden. Geeignete bildgebende Verfahren, vor allem die Magnetresonanztomographie, zeigen eine symmetrische, flächige Degeneration der weißen Substanz mit Kontrastmittelaufnahme. Vorrangig betroffen sind dabei die Occipitallappen, der hintere Bereich des Corpus callosum, die Pyramiden- und die Hörbahn.[3][4]
Therapie
Die therapeutischen Möglichkeiten beschränken sich hauptsächlich darauf, die Symptome der Erkrankung zu lindern. So werden Medikamente gegen spastische Muskelkrämpfe verabreicht, ebenso wie Steroidhormone gegen die neurologischen Begleiterscheinungen. Interferon und Lovastatin bewirken nur selten eine Unterdrückung der entzündlichen Prozesse im Gehirn. Zur Erhöhung der Peroxisomenzahl wird die Gabe von 4-Phenylbutyrat erwogen.[5]
Die einzige kurative Therapie ist die Knochenmarktransplantation, die besonders effektiv ist, wenn sie bei der zerebralen Form in frühen Stadien der Neurodegeneration oder präsymptomatisch erfolgt. Der Mechanismus ist bisher nicht verstanden, es wird aber ein Austausch der myeloischen Zellen durch solche des Spenders vermutet, zu denen möglicherweise auch die Mikroglia des zentralen Nervensystems gehört. Bei HLA-identischen Spendern und frühzeitiger Stammzelltransplantation sind die langfristigen Ergebnisse gut.[2]
Forschung
Nachdem eine französische Arbeitsgruppe um N. Cartier et al. 2009 in einer Proof-of-Concept-Studie zeigten, dass eine autologe Transplantation von CD34-positiven hämatopoetischen Vorläuferzellen möglich ist, die ex vivo mit einem lentiviralen Vektor transfiziert wurden, und dadurch eine Kopie des ABCD1-Gens eingebaut werden konnte,[6] konnte eine offene multizentrische Phase-2-3-Studie 2017 die Sicherheit und Wirksamkeit dieser als Lenti-D (Elivaldogene Tavalentivec) bezeichneten Gentherapie nachweisen.[2] Behandelt wurden 17 Jungen mit im Median 6 Jahren. Zwei Kinder starben im Lauf der Therapie, die übrigen 15 Jungen waren nach mittleren 29 Monaten Nachbeobachtungszeit frei von neurologischen Behinderungen mit nur minimalen klinischen Symptomen. Anders als bei der allogenen Stammzelltransplantation wurde keine Graft-versus-Host-Reaktion beobachtet.
Lorenzos Öl
Einigen Patienten wird als diätische Maßnahme Lorenzos Öl gegeben, eine Mischung aus Glycerin-Trioleat und Glycerin-Trierukat im Verhältnis 4 zu 1. Hypothetisch könnte die Zufuhr dieser langkettigen, einfach ungesättigten Fettsäuren dazu führen, dass die Enzyme, die die überlangkettigen Fettsäuren produzieren, ausgelastet sind und die Konzentration der Fettsäuren auf ein verträglicheres Maß sinkt.[7][8]
Als erster Verfechter der Öl-Therapie gilt Augusto Odone, der Vater des von X-ALD betroffenen Lorenzo. Der medizinische Laie sah sich gezwungen, die Suche nach einem Heilmittel selbst in die Hand zu nehmen, nachdem ihm die behandelnden Ärzte keine großen Hoffnungen auf das Überleben seines Sohnes machen konnten. Entscheidende Anstöße gab der aus der Schweiz stammende Neurologe Hugo Moser, der mit seiner Arbeitsgruppe aus Baltimore eine Studie mit Lorenzos Öl an 89 Jungen durchführte.[9] Die Kinder waren asymptomatisch und bei Behandlungsbeginn im Mittel 4,7 Jahre alt. Nach mittleren 6,9 Jahren Nachuntersuchung zeigten 24 % Veränderungen nur in kernspintomografischen Aufnahmen des Gehirns, 11 % entwickelten kernspintomografische Auffälligkeiten und neurologische Störungen. Allerdings wurden verschiedene Phänotypen zusammen genommen und nicht differenziert, so dass unklar ist, ob und wie viele der Kinder eine zerebrale Adrenoleukodystrophie aufwiesen, die eine sehr schwere Verlaufsform darstellt.
Medien
- Die Geschichte von Lorenzos Öl um die Familie Odone wurde 1992 mit Susan Sarandon, Nick Nolte und Peter Ustinov verfilmt.
- Auch Phil Collins ehrte Lorenzos Überlebenswillen und die Erfolge seines Vaters, der auch ein Projekt zur Erforschung der Myelinisierung von Nervenzellen ins Leben gerufen hat, in dem 1996 veröffentlichten Song Lorenzo auf dem Album Dance into the Light.
- Begleitung des an Adrenoleukodystrophie erkrankten Keno und seiner Mutter über einen Zeitraum von 6 Jahren bis zum Tod des Jungen am 24. Dezember 2017.[10]
Synonyme
- Addison-Schilder-Syndrome
- Fanconi-Prader-Syndrom
- Siemerling-Creutzfeldt-Syndrom
- Adrenoleukomyeloneuropathie (ALMN)
- Adrenomyeloneuropathie (AMN)
Siehe auch
Literatur
- H. W. Moser, A. Mahmood, G. V. Raymond: X-linked adrenoleukodystrophy. In: Nature clinical practice. Neurology. Band 3, Nummer 3, März 2007, S. 140–151, ISSN 1745-8358. doi:10.1038/ncpneuro0421. PMID 17342190. (Review).
- Florian Eichler, Christine Duncan, Patricia L. Musolino, Paul J. Orchard, Satiro De Oliveira, Adrian J. Thrasher, Myriam Armant, Colleen Dansereau, Troy C. Lund, Weston P. Miller, Gerald V. Raymond, Raman Sankar, Ami J. Shah, Caroline Sevin, H. Bobby Gaspar, Paul Gissen, Hernan Amartino, Drago Bratkovic, Nicholas J.C. Smith, Asif M. Paker, Esther Shamir, Tara O’Meara, David Davidson, Patrick Aubourg, David A. Williams: Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy New England Journal of Medicine 2017, Band 377, Ausgabe 17 vom 26. Oktober 2017, Seiten 1630–1638, [DOI: 10.1056/NEJMoa1700554]
- Z. Patay: Diffusion-weighted MR imaging in leukodystrophies. In: European radiology. Band 15, Nummer 11, November 2005, S. 2284–2303, ISSN 0938-7994. doi:10.1007/s00330-005-2846-2. PMID 16021451. (Review).
- J. H. Kim, H. J. Kim: Childhood X-linked adrenoleukodystrophy: clinical-pathologic overview and MR imaging manifestations at initial evaluation and follow-up. In: Radiographics : a review publication of the Radiological Society of North America, Inc. Band 25, Nummer 3, 2005 May-Jun, S. 619–631, ISSN 1527-1323. doi:10.1148/rg.253045118. PMID 15888613. (Review).
- C. Gondcaille, M. Depreter u. a.: Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. In: Journal of Cell Biology. Band 169, Nummer 1, April 2005, S. 93–104, ISSN 0021-9525. doi:10.1083/jcb.200501036. PMID 15809314. PMC 2171887 (freier Volltext).
- N. Cartier, S. Hacein-Bey-Abina, C. C. Bartholomae et al.: Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy Science 2009, Band 326, Seiten 818–823.
- E. Simon: Efficacy of Lorenzo oil in adrenomyeloneuropathy. In: Annals of neurology. Band 36, Nummer 1, Juli 1994, S. 116–117, ISSN 0364-5134. doi:10.1002/ana.410360126. PMID 8024253.
- H. W. Moser: Lorenzo oil therapy for adrenoleukodystrophy: a prematurely amplified hope. In: Annals of neurology. Band 34, Nummer 2, August 1993, S. 121–122, ISSN 0364-5134. doi:10.1002/ana.410340202. PMID 8338333.
- H. W. I. Moser, G. V. Raymond, S. E. Lu, L. R. Muenz, A. B. Moser, J. Xu, R. O. Jones, D. J. Loes, E. R. Melhem, P. Dubey, L. Bezman, N. H. Brereton, A. Odone: Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's oil Archives of Neurology 2005, Band 62, Ausgabe 7 vom Juli 2005, Seiten 1073–80.
- Kenos kurzes Leben. Abgerufen am 8. Februar 2019.