Morbus Wilson
Die Wilson-Krankheit oder der Morbus Wilson (weitere Synonyme: Wilsonsche Krankheit, Pseudosklerose Westphal, Westphal-Strümpellsche Pseudosklerose und hepatolentikuläre Degeneration (lateinisch Degeneratio hepatolenticularis)) ist eine autosomal-rezessiv vererbte Erkrankung, bei der durch eine oder mehrere Genmutationen der Kupferstoffwechsel in der Leber gestört ist. In ihrer Folge kommt es bei dieser Kupferspeicherkrankheit zu einer verminderten Kupferausscheidung über die Galle, woraus eine vermehrte Ansammlung von Kupfer in der Leber, dem Auge, dem Zentralnervensystem und anderen Organen resultiert. Daraus ergibt sich ein vielgestaltiges Muster von Symptomen, das sich vor allem in Leberschäden und neurologischen Defiziten äußert. Die Krankheit ist durch Medikamente, die den Kupferspiegel im Blut senken oder die Aufnahme des Kupfers verhindern, gut zu behandeln. Als letzte Alternative steht die Lebertransplantation zur Verfügung. Die Krankheit wurde nach dem britischen Neurologen Samuel Alexander Kinnier Wilson benannt.
| Klassifikation nach ICD-10 | |
|---|---|
| E83.0 | Morbus Wilson |
| ICD-10 online (WHO-Version 2019) | |
Angeborene Kupferspeicherkrankheiten kommen häufiger auch beim Haushund vor, hier vor allem beim Bedlington-Terrier (→ Angeborene Kupferspeicherkrankheit des Hundes).
Die Bezeichnung hepatolentikuläre Degeneration für Morbus Wilson gilt als veraltet, da Schäden durch Kupferansammlung nicht nur im Linsenkern (Nucleus lentiformis) vorzufinden sind, sondern im gesamten Gehirn. Die Linsenkerndegeneration (im Englischen Wilson’s syndrome) und die Pseudosklerose Westphal wurden auch als eine Einheit bildende Syndrome angesehen.[1]
Häufigkeit
Die Inzidenz wird populationsabhängig zwischen 1:30.000 und 1:300.000 Einwohnern angegeben.[2] Das mutierte Gen ist weltweit verbreitet, etwa einer von hundert gesunden Menschen ist heterozygot und kann somit potenziell die Krankheit an seine Kinder weitergeben.[3] Aufgrund des autosomal-rezessiven Erbgangs ergibt sich ein statistisches Risiko von 1:4 für Geschwister einer erkrankten Person ebenfalls zu erkranken. Für Kinder eines erkrankten Patienten ergibt sich ein Risiko von 1:200.[4]
Genetik
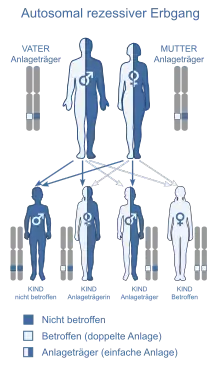
Die Ursache der Erkrankung ist eine Mutation im Gen ATP7B, dem „Wilson-Gen“, das sich auf Chromosom 13 (Genort 13q14.3) befindet. Dieses codiert das „Wilson-Protein“, eine Kupfer bindende, Kationen transportierende ATPase. Es sind über 370 verschiedene Mutationen des 21 Exons umfassenden Wilson-Gens bekannt, was den unterschiedlichen Verlauf der Wilson-Krankheit erklärt und die genetische Diagnose erschwert. Die Häufigkeit verschiedener Mutationen im Wilson-Gen wird durch seine im Vergleich zu anderen Genen große Länge erklärt. Die meisten Patienten, die an der Krankheit leiden, weisen auf ihren beiden Chromosomen jeweils zwei unterschiedliche Mutationen des Gens auf.[3][5] Am häufigsten (50 bis 80 %) ist in Mitteleuropa die Punktmutation His1069Gln in Exon 14.
Pathophysiologie
Durch die Mutation entsteht eine Fehlleistung des Wilson-Proteins in den Leberzellen, das unter anderem für den Transport von Kupfer aus der Leber in die Galle verantwortlich ist. Beim Morbus Wilson wird das Kupfer nicht, wie bei einem gesunden Menschen üblich, mit der Galle und somit über den Stuhl ausgeschieden, sondern im Organismus eingelagert und entfaltet in verschiedenen Organen seine toxische Wirkung. Der genaue Mechanismus, durch den der hohe Kupferspiegel die Zellen schädigt, ist noch nicht abschließend geklärt. Man vermutet, dass der Überschuss an Kupfer die Bildung von Radikalen innerhalb der Zellen fördert.[3]
Der primäre Anreicherungsort des Kupfers ist die Leber, ein weiteres häufig betroffenes Organ ist das Auge. Bei rund 45 % der Patienten ist auch das zentrale Nervensystem betroffen. Bei rund 15 % werden die roten Blutzellen durch das Kupfer geschädigt. Selten ist die toxische Anreicherung in den Nieren und im Herzmuskel.[5]
Altersverteilung
Häufig manifestiert sich die Krankheit im zweiten oder dritten Lebensjahrzehnt. Der Zeitpunkt ist allerdings sehr variabel. Es sind auch Fälle beschrieben, bei denen sich erste Symptome bereits in der Kindheit ab dem sechsten Lebensjahr gezeigt haben. Erstmanifestationen jenseits des 32. Lebensjahres sind selten, doch sind auch Patienten beschrieben, bei denen erst im hohen Alter Erscheinungen der Krankheit fassbar wurden.[5][6] In einem Fallbericht wurde auf einen zweijährigen Patienten verwiesen, bei dem die Erkrankung aufgrund abnormaler Laborwerte bereits im frühesten Kindesalter diagnostiziert werden konnte.[7]
Symptome
Leber
Die Leber ist sehr häufig geschädigt, da die Leberzellen der primäre Ort der Kupferspeicherung sind und somit als erste von der Überladung betroffen werden. Das Ausmaß des Leberschadens ist jedoch sehr variabel. Das Spektrum reicht von einer asymptomatischen Erhöhung der Transaminasen oder einer Lebervergrößerung bis zu einer schnellen und dabei auch sehr schwer (fulminant) verlaufenden Leberentzündung (Hepatitis) mit lebensbedrohlichem Verlauf. Der klassische Verlauf ist allerdings chronisch, es bildet sich unbehandelt über eine Leberverfettung schließlich eine Leberzirrhose aus, die letztlich im Versagen des Organs endet.[5][8] Der Morbus Wilson geht jedoch nicht mit einem erhöhten Risiko einher, an Leberzellkrebs zu erkranken.[3] Durch einen massiven Untergang von Leberzellen können große Mengen an Kupfer freigesetzt werden, was zu einer fulminant verlaufenden Hepatitis führen kann.[5] Bei rund 5 % der Patienten führt ein fulminantes Leberversagen zur Erstdiagnose, ohne dass die Krankheit vorher erkannt wurde.[9]
Augen
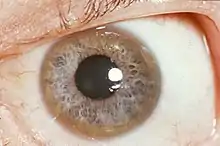
Häufig sind ebenso Symptome an den Augen der erkrankten Menschen. Der Kayser-Fleischer-Kornealring zeigt sich als goldbrauner bis grünlicher Rand, der die Iris umgibt. Er kommt durch die Einlagerung von Kupfer in die Hornhaut zustande[5] und kommt bei 95 % der Patienten mit neurologischen Symptomen, allerdings bei nur etwas über 50 % der Patienten mit primärer Leberbeteiligung vor.[10] Nachtblindheit kann ebenso durch die Kupfereinlagerung ausgelöst werden.[8] Durch eine augenärztliche Untersuchung kann auch bei einem Teil der Patienten eine Sonnenblumen-Katarakt festgestellt werden.[9] Darunter versteht man gelb-braune Kupferablagerungen in der Augenlinse,[11] die aber nicht zu einer Beeinträchtigung der Sehleistung führen. Ebenso kann die Erkrankung Schielen oder auch eine Entzündung des Sehnervs auslösen.[9]
Zentrales Nervensystem
Das Hauptkrankheitsanzeichen (Kardinalsymptom) am zentralen Nervensystem ist eine Bewegungsstörung, die auch als flapping tremor bezeichnet wird: Die Erkrankung verursacht sehr variable, teilweise parkinsonähnliche oder auch Chorea-artige Symptome wie unwillkürliche ruckartige Zuckungen oder Zittern der Extremitäten. Der Tonus der Muskulatur kann verringert oder erhöht (Rigor) sein. Ebenso kann sie sich in einer verwaschenen Sprache äußern.[5] Auch können Koordinationsstörungen, Störungen der Feinmotorik, muskuläre Verkrampfungen und Schluckstörungen auftreten. Als mögliche vegetative Störung ist erhöhter Speichelfluss beschrieben. Selten sind Spastiken und epileptische Anfälle. Zu diesen vielgestaltigen neurologischen Symptomen gesellen sich in insgesamt 10 % der Fälle noch eine Vielzahl möglicher psychiatrischer Beeinträchtigungen. Der Morbus Wilson kann hierbei Verminderungen der Intelligenzleistung, einer subkortikalen Demenz,[12][13] Beeinträchtigungen des sozialen Umgangs sowie Depressionen und auch Psychosen auslösen.[8]
Seltenere Symptome
Durch einen massiven Untergang von Leberzellen können große Mengen an Kupfer freigesetzt werden, was die Blutzellen schädigen und zu einer hämolytischen Anämie führen kann. Selten sind Schädigungen der Niere und des Herzmuskels. Der Nierenschaden kann unbehandelt in ein nephrotisches Syndrom übergehen und bis zum Nierenversagen fortschreiten. Am Herzmuskel kann die Krankheit eine Kardiomyopathie verursachen.[5] Zusätzlich zu diesen eher seltenen Manifestationsorten sind auch Schädigungen des Knochens beschrieben. So kann der Morbus Wilson Osteomalazie und Osteoporose verursachen oder fördern.[3]
Diagnose
Die Diagnose eines Morbus Wilson ist nicht immer einfach zu stellen. Die Gründe hierfür sind einerseits die Seltenheit der Erkrankung und andererseits die Vielfalt der möglichen Symptome. Insbesondere bei Patienten im Kindes- und Jugendalter sollte bei unklarer Leberwerterhöhung und nicht eindeutig erklärbaren neurologischen Symptomen ein Morbus Wilson ausgeschlossen werden.
Die technisch einfachste Untersuchung ist die Inspektion des Auges mittels Spaltlampe. Der Kayser-Fleischer-Ring ist dabei ein auffälliges Zeichen der Krankheit, er ist allerdings nicht bei allen Patienten nachweisbar; bei dominierender neurologischer Symptomatik ist er fast zwingend (obligat), bei vorwiegend hepatischen Symptomen weitaus seltener.
Laboruntersuchungen
Ebenso wegweisend, aber nicht bei allen Patienten vorhanden, sind typische Laborwertveränderungen. Das transportierende Protein Coeruloplasmin ist dabei im Serum als Folge des gestörten Kupferhaushaltes erniedrigt. Dabei ist zu beachten, dass Coeruloplasmin ein Akute-Phase-Protein ist und somit im Rahmen von Entzündungen einen falsch-hohen Wert liefern kann. Der Gesamtkupfergehalt im Blut ist oft erniedrigt. Der Anteil des freien Kupfers jedoch häufig erhöht. Die Kupferwerte im Urin sind oft erhöht. Ferner ist der Coombs-Test in Zusammenhang mit einer möglichen hämolytischen Anämie ein weiterer Indikator.[14] Bei unauffälligem Labor empfiehlt sich der Penicillamintest. Hierbei wird das freie Kupfer im Urin nach Gabe von 500 mg Penicillamin gemessen. Der Test ist jedoch nur für Kinder standardisiert, was seine Aussagekraft bei Erwachsenen deutlich einschränkt. Liegt der Kupferwert im Sechs-Stunden-Sammelurin über 600 µg, weist der Test auf das Vorhandensein eines Morbus Wilson hin. Bei nicht eindeutigen Testergebnissen kann der Radiokupfertest durchgeführt werden. Dabei wird der Einbau radioaktiv markierten Kupfers in das Coeruloplasmin über 48 Stunden überwacht.[5][9][15] Zur Gesamtanalyse aller per se nicht eindeutigen Laboruntersuchungen wird häufig das Scoring-Modell der 8. internationalen Morbus-Wilson-Konferenz verwendet, das alle Kann-Ergebnisse summiert und somit eine vergleichsweise valide Diagnose ermöglicht.[16]
Leberbiopsie
Als invasive diagnostische Maßnahme kann eine Leberbiopsie durchgeführt werden. Bei einem Kupfergehalt von mehr als 250 µg/g Lebergewebe und einem erniedrigten Coeruloplasmin ist von einem Morbus Wilson auszugehen. Erhöhte Kupferwerte in der Leber finden sich aber auch bei anderen Erkrankungen dieses Organs, zum Beispiel der Primär biliären Zirrhose. Auch kann der Kupferwert in einer zirrhotisch umgebauten Leber falsch negativ ausfallen, da der Gehalt von kupferspeichernden Leberzellen zu Gunsten von Bindegewebszellen vermindert ist. Trotz dieser Fehlerquellen gilt die Leberbiopsie als Goldstandard bei der Diagnose des Morbus Wilson.[5][9]
Neurologische und neuroradiologische Untersuchungen
Bei gesicherter Diagnose eines Morbus Wilson werden eine neurologische Untersuchung und eine MR-Untersuchung des zentralen Nervensystems empfohlen. Die Wiederholung der Untersuchungen im Verlauf ermöglicht eine Erfolgskontrolle der Therapie.[17]
Molekulargenetische Untersuchungen
Es können mehrere Genloci für den Morbus Wilson verantwortlich sein. Für den häufigsten Genort sind mittlerweile rund 300 Mutationen beschrieben worden. Zur Krankheitsauslösung kommt es häufig durch Compoundheterozygotie. Homozygote Fälle sind sehr selten. Ebenso kann häufig die Korrelation zwischen genetischen Veränderungen und klinischer Ausprägung deutlich variieren.[17]
Pathologie
In manchen Fällen lässt sich die krankheitsbedingende Kupfereinlagerung in der Leber bereits in einem frühen Stadium feingeweblich (histologisch) erkennen. Dabei eingesetzte Färbungen, die Kupfer sichtbar machen, sind die Rhodanin- oder Rubeansäurefärbung. Da der Nachweis aber auch häufig trotz Vorhandensein der Erkrankung fehlschlagen kann, sind Kupferfärbungen von begrenztem diagnostischen Wert.[9][4]
In einem fortgeschrittenen Stadium zeigen sich unter dem Mikroskop vergrößerte Leberzellen, die oft Glykogeneinschlüsse in den Zellkernen aufweisen. Oft zeigt sich zusätzlich ein der Hepatitis ähnliches Bild mit Lymphozyten, die die Bindegewebsfasern (Septen) und Portalfelder, in welchen Gefäße und Gallengänge verlaufen, der Leber infiltrieren. Das histologische Bild ist somit auch im fortgeschrittenen Stadium nicht spezifisch für die Krankheit und erlaubt keine zuverlässige Diagnose.[18] Im Endstadium der Leberschädigung geht das Bild in eine Zirrhose über. Diese kann kleinknotig oder gemischt klein- und großknotig erscheinen. In 50 % der Fälle sind Mallory-Körper nachweisbar, die auch bei einer alkoholischen Leberschädigung vorkommen.[9]
Im Gehirn zeigt sich unter dem Mikroskop eine Vermehrung der Gliazellen mit schwammartiger Auflockerung des Gehirngewebes. Typisch (aber nicht für die Erkrankung spezifisch) sind die nach dem Entdecker, dem polnischen Neurologen Adam Opalski (1897–1963), benannten sogenannten Opalski-Zellen, bei denen es sich um degenerierte Astrozyten mit granulärem Zytoplasma handelt.[19]
Therapie
Diät
Eine kupferarme Diät ist schwierig durchzuführen, da Kupfer in sehr vielen Lebensmitteln vorkommt. Selbst bei maximaler Einhaltung von Diätregeln ist sie nicht als einzige Behandlung zu empfehlen. Empfehlenswert im Rahmen der Diät ist es, den Verzehr von Leber, Niere, Hirn, Schokolade, Kakao, Nüssen, Pilzen, Bohnen, Rosinen und Krustentieren zu vermeiden.[5]
Chelatbildner
Zur medikamentösen Therapie werden in erster Linie Chelatbildner verwendet. Diese Medikamente bilden mit Kupfer Chelatkomplexe und fangen es somit aus dem Blut. Die wasserlöslichen Komplexe werden mitsamt dem Kupfer über die Nieren in den Urin ausgeschieden. Es stehen zwei Medikamente zur Verfügung – D-Penicillamin und Trientin. Welches Medikament die erste Wahl darstellen soll, ist bisher umstritten.[9]
Penicillamin wird in der Leitlinie der Deutschen Gesellschaft für Neurologie von 2005 über den Morbus Wilson weiterhin als Mittel der ersten Wahl empfohlen.[8] Die den Einsatz von Penicillamin am meisten begrenzende Nebenwirkung ist, dass sich in 20 % der Fälle die neurologische Symptomatik verschlechtert.[5] Aus diesem Grunde wird in der anglo-amerikanischen Literatur der Einsatz von Penicillamin als Mittel der ersten Wahl bei Patienten mit neurologischer Symptomatik nicht empfohlen.[3] Um dieses Problem zu umgehen, empfiehlt die Leitlinie die einschleichende Aufdosierung des Medikaments, da die neurologische Verschlechterung auf anfänglich durch die Therapie erhöhte Kupferspiegel im Blut zurückzuführen sei.[8] Für diese Praktik gibt es allerdings bis heute keine Evidenz.[9] Weitere mögliche Nebenwirkungen der Penicillamintherapie sind Hörstörungen, Hautreaktionen, Fieber, Nierenschäden und Lupus erythematodes. Des Weiteren wirkt Penicillamin als Antagonist zu Vitamin B6, so dass eine Substitution des Vitamins angebracht ist.[5] Penicillamin ist bei Schwangerschaft nicht anzuwenden und tritt auch in die Muttermilch über.[8]
Als alternativer Chelatkomplexbildner kann Trientin eingesetzt werden. Es wird in der anglo-amerikanischen Literatur mittlerweile als Medikament der ersten Wahl empfohlen. Laut diesen Autoren böte es ein günstigeres Nebenwirkungsprofil und löse nicht so häufig wie Penicillamin neurologische Verschlechterungen aus.[4] Eine seltene Nebenwirkung ist die Panzytopenie. Ebenso wirkt das Medikament fruchtschädigend. Im Gegensatz zu Penicillamin sind bisher keine Nierenschäden und Hypersensitivitätsreaktionen berichtet worden. Trientin wird aber eine schwächere kupferbindende Wirkung als Penicillamin nachgesagt. Da bisher keine randomisierten Studien vorliegen, die beide Medikamente direkt miteinander vergleichen, ist dieses Thema bis heute umstritten.[9]
Unstrittig ist jedenfalls, dass beide Chelatbildner rund eine bis zwei Stunden nach dem Essen eingenommen werden müssen, da die Aufnahme der Medikamente gleichzeitig mit der Nahrung unzureichend ist.[9]
Kupferaufnahmehemmer
Ebenso kommen Zinkpräparate in Frage. Durch Zink wird der Stoffwechsel der Darmzellen so verändert, dass weniger Kupfer aufgenommen wird. Zink verursacht im Gegensatz zu den Chelatbildnern keine Schädigungen an ungeborenen Kindern und kann deshalb während Schwangerschaft und Stillzeit eingesetzt werden.[8] Bei 10 % der Patienten treten eine Refluxösophagitis (Sodbrennen) oder Übelkeit als Nebenwirkungen auf.[4]
In den USA und Kanada befindet sich ein weiterer Hemmer der Kupferaufnahme namens Tetrathiomolybdat zurzeit in Erprobung.[3] Der Wirkstoff bildet im Blut zusammen mit Albumin und frisch aufgenommenem Kupfer Komplexe, die von der Leber verstoffwechselt und über die Galle ausgeschieden werden. Eine erste randomisierte Studie zeigte im Vergleich zu Trientinen ein günstigeres Nebenwirkungsprofil bezüglich der Verschlechterung neurologischer Symptome.[9]
Verlaufsbeobachtung und Therapieregime
Der Therapieerfolg mit Chelatbildnern kann durch die Bestimmung des Kupfers im 24-Stunden-Sammelurin überwacht werden. Bei Kupferaufnahmehemmern kann man den Erfolg der Behandlung über den Kupfergehalt des Stuhls verfolgen. Die Compliance des Patienten kann bei Zinkpräparaten über den Zinkgehalt des Urins festgestellt werden. Des Weiteren ist die Bestimmung von Proteinen im Urin anzuraten um den Verlauf der Nierenfunktion verfolgen zu können.[5][9] Der Verlauf des Leberschadens ist durch Ultraschalluntersuchungen und die Bestimmung der Transaminasen überwachbar. Um den Verlauf der neurologischen Schädigungen zu verfolgen, ist die neurologische Untersuchung unter Zuhilfenahme eines Scores angebracht. Bildgebende Verfahren (Computertomographie, Magnetresonanztomografie) sowie die Elektroenzephalografie können auch Hinweise auf den Verlauf der Erkrankung unter Therapie geben.[8] Im Zuge des Therapieverlaufs ist darauf zu achten, dass der Kupferspiegel nicht unter das Bedarfsniveau des Körpers absinkt. Erste Anzeichen eines Kupfermangels sind Blutarmut oder eine Verminderung der Zahl der weißen Blutzellen (Leukopenie).[4]
Je nach Erfolg der Therapie kann diese individuell angepasst werden. Manche Autoren sprechen sich dafür aus, nach dem Entleeren der Kupferspeicher mit Chelatbildnern allein auf Zinkpräparate zu setzen. Wiederum andere sprechen sich für eine Kombinationsbehandlung aus beiden aus.[8] Einige Autoren halten auch eine lebenslange Therapie mit Chelatbildnern für sinnvoll.[5]
Lebertransplantation
Bei schweren Verläufen mit einer starken Leberschädigung kann eine Transplantation des Organs angestrebt werden. Die Krankheit wird dadurch geheilt, da der Patient die gesunden Leberzellen ohne Gendefekte vom Spenderorgan erhält.[5] In einer krisenhaften Verschlimmerung der Krankheit durch exzessiv hohe Kupferspiegel kann, bis eine Transplantation möglich gemacht wurde, notfallmäßig freies Kupfer durch die Infusion von Albumin gebunden werden.[8]
Asymptomatische Patienten
Ist bei einem Patienten die Diagnose Morbus Wilson gestellt, so ist ein Screening in seiner Familie auf noch asymptomatische Erkrankte notwendig. Es sollte die Geschwister und auch die Kinder des Patienten einschließen. Da eine Suche nach den über 250 Mutationen nicht praktikabel ist, wird eine Haplotypanalyse durchgeführt. Dabei wird nicht nach den über 250 Mutationen gesucht, sondern es wird nachgewiesen, ob die Angehörigen die entsprechenden Chromosomenbereiche, die Mutationen tragen, von ihren Eltern geerbt haben. Sollte ein für die Krankheit Homozygoter auf diese Weise festgestellt werden, ohne dass er Symptome zeigt, ist eine Behandlung mit Zinkpräparaten angezeigt (indiziert). Dadurch soll verhindert werden, dass sich der Körper des betreffenden Menschen überhaupt erst mit Kupfer aufsättigen kann.[8]
Prognose
Unbehandelt führt der Morbus Wilson bei frühem Auftreten (in der Kindheit) und vorrangig internistischen Komplikationen (Leberversagen, Nierenversagen, Hämolyse) binnen 2 bis 7 Jahren zum Tod; die Lebenserwartung ist bei den später einsetzenden vorwiegend neurologisch oder psychiatrisch auffälligen Patienten günstiger (über 10 bis 40 Jahre schleichender Verlauf).
Frühzeitig erkannt und lebenslang therapiert ist der Morbus Wilson als gut behandelbar anzusehen. Die Lebenserwartung unterscheidet sich dann nicht von gesunden Menschen. Unbehandelt oder als schwerer Morbus Wilson verläuft die Krankheit oft tödlich.[5] Neurologische Ausfälle können durch die Therapie geheilt werden, sofern sie nicht bereits mehrere Jahre bestehen. Falls die Schädigung der Leber nicht bereits zu einer Leberzirrhose geführt hat, ist auch dieser Schaden durch die Therapie beeinflussbar.[8]
Bei rund drei Vierteln der Patienten kann das Fortschreiten der Erkrankung aufgehalten oder ein Rückgang der Symptome erzielt werden. Patienten, die primär an neurologischen Symptomen litten, haben dabei ein schlechteres Outcome als Patienten, bei denen die Leberschädigung führend ist.[20]
Forschungsgeschichte
Entdeckt und beschrieben wurde die unter verschiedenen Namen bekannte Krankheit[21] erstmals 1854 von Friedrich Theodor von Frerichs. Eine detailliertere Darstellung erfolgte 1898 von Carl Friedrich Otto Westphal sowie Adolf von Strümpell. Die heute favorisierte Namensgebung erfolgte durch die umfassende Darstellung durch Samuel Alexander Kinnier Wilson in seiner Doktorarbeit, für die er 1912 eine Auszeichnung erhielt. Die Augenärzte Bernhard Kayser (1869–1954) und Bruno Fleischer beschrieben Kupferablagerungen in der Hornhaut des Auges (Kayser-Fleischer-Kornealring). 1948 identifizierte John Nathaniel Cumings (1906–1974) eine Störung des Kupferstoffwechsels als Ursache.[22]
Der erste Therapieversuch mit einem Chelatbildner erfolgte 1951 mit 2,3-Dimercaptopropanol. Bis dahin führte die Krankheit in den meisten Fällen zum Tod des Patienten. 1956 wurde Penicillamin als wirksam beschrieben und ersetzte das ältere Medikament, da Penicillamin wirksamer und weniger nebenwirkungsbehaftet ist.[8][23] Dass die Krankheit eine hämolytische Anämie verursachen kann, wurde 1967 festgestellt.[24] 1969 wurde Triethylentetramin als alternativer Chelatbildner eingeführt. Ebenso begann in den 1960er Jahren die Therapie mit Zinkverbindungen.[9]
Das betroffene Gen ATP7B wurde 1993 von mehreren unabhängigen Forschungsgruppen auf dem langen Arm des Chromosom 13 (13q14.3) lokalisiert.[9]
Literatur
Leitlinien
- S1-Leitlinie Morbus Wilson der Deutschen Gesellschaft für Neurologie (DGN). In: AWMF online (Stand 2012)
Lehrbücher
- Anthony Fauci u. a.: Harrison's Principles of Internal Medicine. Band 2, New York 2008.
- Herbert Renz-Polster, Steffen Krautzig: Basislehrbuch Innere Medizin. 4. Auflage. München 2008
- Andreas Straube, Wieland Hermann: Morbus Wilson. In: Thomas Brandt, Johannes Dichgans, Hans Christoph Diener (Hrsg.): Therapie und Verlauf neurologischer Erkrankungen. 5. Auflage. Kohlhammer, Stuttgart 2007, ISBN 978-3-17-019074-0.
- Raphael Rubin, David Strayer u. a.: Rubin's Pathology. 5. Auflage. Philadelphia 2008.
- Ursus-Nikolaus Riede: Allgemeine und Spezielle Pathologie. Stuttgart 2004.
Artikel in Fachzeitschriften
- A. Ala, A. P. Walker, K. Ashkan, J. S. Dooley, M. L. Schilsky: Wilson's disease. In: The Lancet. Band 369, Nummer 9559, Februar 2007, S. 397–408, ISSN 1474-547X. doi:10.1016/S0140-6736(07)60196-2. PMID 17276780. (Review).
- U. Merle, M. Schaefer, P. Ferenci, W. Stremmel: Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. In: Gut. Band 56, Nummer 1, Januar 2007, S. 115–120, ISSN 0017-5749. doi:10.1136/gut.2005.087262. PMID 16709660. PMC 1856673 (freier Volltext).
- R. F. Pfeiffer: Wilson's Disease. In: Seminars in neurology. Band 27, Nummer 2, April 2007, S. 123–132, ISSN 0271-8235. doi:10.1055/s-2007-971173. PMID 17390257. (Review).
- G. J. Brewer, F. K. Askari: Wilson's disease: clinical management and therapy. In: Journal of Hepatology. Band 42 Suppl, Nummer 1, 2005, S. S13–S21, ISSN 0168-8278. doi:10.1016/j.jhep.2004.11.013. PMID 15777568. (Review).
Weblinks
- Morbus Wilson. In: Orphanet (Datenbank für seltene Krankheiten).
- Morbus Wilson. In: Online Mendelian Inheritance in Man. (englisch)
- Morbus Wilson e.V. Deutscher Selbsthilfe-Verein für Betroffene
- EuroWilson Morbus Wilson Informations-Seite von europäischen Ärzten, unterstützt von der Europäischen Kommission
- University of Alberta Morbus Wilson Datenbank genetischer Mutationen
Einzelnachweise
- Immo von Hattingberg: Hepatolentikuläre Degeneration. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 1345 f.
- Olsson u. a.: Determination of the frequencies of ten allelic variants of the Wilson disease gene (ATP7B), in pooled DNA samples. In: Eur J Hum Genet. 2000;8(12), S. 933–938. PMID 11175281
- R. Rubin, E. Rubin: The Liver and Biliary System. In: R. Rubin, D. Strayer u. a.: Rubin's Pathology. 5. Auflage. Philadelphia 2008, S. 653f.
- G. Brewer: Wilson Disease. In: A. Fauci u. a.: Harrison's Principles of Internal Medicine. Band 2, New York 2008, S. 2449–2552.
- H. Renz-Polster, S. Krautzig: Basislehrbuch Innere Medizin. 4. Auflage. München 2008, S. 921–923.
- J. Trojanowski, L. Kenyon, T. Bouldin: The Nervous System. In: R. Rubin, D. Strayer u. a.: Rubin's Pathology. 5. Auflage. Philadelphia 2008, S. 1214.
- A. Beyersdorff, A. Findeisen: Morbus Wilson: Case report of a two-year-old child as first manifestation. In: Scandinavian Journal of Gastroenterology. 2006 Apr;41(4), S. 496–497; PMID 16635921
- Leitlinie der Deutschen Gesellschaft für Neurologie zum Morbus Wilson erstellt 2003, aktualisiert 2005, online abrufbar als html zuletzt abgerufen am 11. September 2008.
- A. Ala, A. Walker, K. Ashkan, J. Dooley, M. Schilsky: Wilson's Disease. In: The Lancet. 2007 Feb 3;369(9559), S. 397–408, PMID 17276780
- European Association for the Study of the Liver: EASL Clinical Practice Guidelines: Wilson's disease. In: Journal of hepatology. Band 56, Nr. 3. ELSEVIER, 2012, S. 671–685, doi:10.1016/j.jhep.2011.11.007 (englisch).
- Atlas of Ophthalmology online, abrufbar als html; Zuletzt abgerufen am 13. Oktober 2008.
- R. M. Bonelli, J. L. Cummings: Frontal-subcortical dementias. In: Neurologist. 2008 Mar;14(2), S. 100–107. PMID 18332839.
- C. Lang, D. Müller, D. Claus, K. F. Druschky: Neuropsychological findings in treated Wilson's disease. In: Acta Neurol Scand. 1990 Jan;81(1), S. 75–81. PMID 2330819.
- EuroWilson: Diagnosis, abgerufen am 22. November 2012.
- Gerd Herold: Innere Medizin. Köln, 2009, S. 516.
- Morbus Wilson Scoring System: http://www.eurowilson.org/professional/diagnosis/index.phtml#Scoring-system
- Eve A. Roberts, Michael L. Schilsky: Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008 Jun;47(6):2089-111. PMID 18506894
- H. Denk, H.P. Dienes, M. Trauner: Leber und intrahepatische Gallenwege. In: Ursus-Nikolaus Riede: Allgemeine und Spezielle Pathologie. Stuttgart 2004, S. 789.
- A. Opalski: Über eine besondere Art von Gliazellen bei der Wilson-Pseudosklerosegruppe. In: Zeitschrift für die gesamte Neurologie und Psychiatrie. (1930) 124(1), S. 420–425.
- U. Merle, M. Schaefer, P. Ferenci, W. Stremmel: Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study. In: Gut. 2007 Jan;56(1), S. 115–120. Epub 2006 May 18; PMID 16709660
- Vgl. auch Ludwig Weissbecker: Die hepatolentikuläre Degeneration (Wilsonsche Krankheit, Westphal-Strümpellsche Pseudosklerose). In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 1114 f.
- J. N. Cumings: The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. In: Brain: a journal of neurology. Band 71, Pt. 4, Dezember 1948, S. 410–415, ISSN 0006-8950. PMID 18124738.
- J. M. Walshe: Wilson's disease; new oral therapy. In: The Lancet. 1956 Jan 7;270(6906), S. 25–26. PMID 13279157
- N. McIntyre, H. M. Clink, A. J. Levi, J. N. Cumings, Sheila Sherlock: Hemolytic anemia in Wilson's disease. In: New England Journal of Medicine. 1967 Feb 23;276(8), S. 439–444, PMID 6018274