Transforming Growth Factor beta
Transforming Growth Factor β (TGFβ) bezeichnet drei Zytokine, die gemeinsam die TGFβ-Familie bilden. In Wirbeltieren gibt es drei TGFβ Formen: TGFβ1, TGFβ2 und TGFβ3 (humane Gennamen: TGFB1, TGFB2, TGFB3). Trotz der Namensähnlichkeit gibt es keine enge strukturelle oder evolutionäre Verwandtschaft mit den TGFα Wachstumsfaktoren[1].
| Transforming Growth Factor β | ||
|---|---|---|
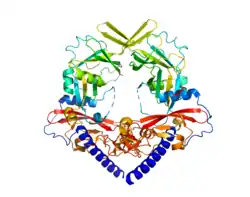
| ||
| PDB ID: 3VI4. Regenbogenfarben, blau N-terminus, rot C-terminus | ||
|
Vorhandene Strukturdaten: PDB 1KLC, PDB 2TGI, PDB 1TGK, PDB 3RJR,PDB 5FFO | ||
| Masse/Länge Primärstruktur | TGFβ1/2/3: Signalpeptid: 29/20/23; LAP: 249/282/277; TGFβ: 111/111/111 | |
| Bezeichner | ||
| Externe IDs | ||
Knockout-Mäuse haben gezeigt, dass
- TGFβ1 das Immunsystem dämpft[2],
- TGFβ2 an der Entwicklung von Lunge, Herz, Knochen und Urogenitaltrakt beteiligt ist[3],
- TGFβ3 ebenfalls zur Lungenentwicklung beiträgt. TGFβ3 Signale sind außerdem wichtig um die Entstehung von Gaumenspalten zu verhindern[4][5].
Alle drei TGFβ scheinen in der Lage, Zellen verschiedenen Ursprungs in Myofibroblasten zu transformieren. Diese zeichnen sich durch erhöhte Kontraktilität und Sekretion von EZM Proteinen aus[6]. Dies sind einerseits Voraussetzungen für eine erfolgreiche Wundheilung, führen aber andererseits zur Narbenbildung, die im pathologischen Fall zur Fibrose und im Extremfall zum Organversagen führt.
TGFβ werden durch Zellen in einer inaktiven Form in der extrazellulären Matrix (EZM) deponiert. Erst durch Aktivierung wird das eigentliche Zytokin TGFβ frei und kann an zelluläre Oberflächenrezeptoren binden[7]. Diese TGFβ-Rezeptoren gehören zur Familie der Serin/Threonin-Kinasen. Aktivierte TGFβ-Rezeptoren aktivieren intrazellulär Proteine der Smad-Familie, die daraufhin in den Zellkern gelangen und mit Kofaktoren die Genexpression beeinflussen[8]. Die drei Vertreter der TGFβ Familie haben spezifische Funktionen in der Entwicklung von Wirbeltieren und andauernde Aktivierung des TGFβ Signalwegs wird mit verschiedenen Krankheiten in Verbindung gebracht.
Struktur
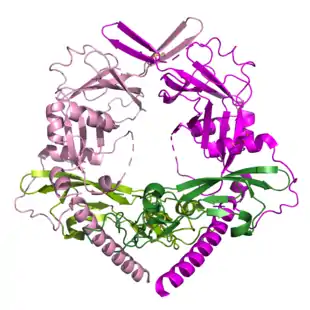
TGFβ1-3 sind nur drei Vertreter der gesamten TGFβ-Familie. Diese besteht aus folgenden Untergruppen:
- bone morphogenetic protein (BMP)
- growth and differentiation factors (GDF)
- activin
- TGFβ
Alle 33 Gene der Vertreter der TGFβ Familie führen zu einem Polypeptid, das in ein Sekretionssignalpeptid, eine Prodomäne (ca. 250 Aminosäuren, etwa 30 kDa) und das eigentliche Zytokin (ca. 110 Aminosäuren, etwa 13 kDa) gespalten werden[1]. Die Spaltung des Polypeptids in den funktionellen Prodomänen-Zytokin-Komplex findet im Golgi-Apparat statt. Anschließend wird dieser Komplex in den extrazellulären Raum sekretiert[9]. Speziell bei TGFβ1-3 ist das Zytokin eng mit der Prodomäne verbunden und dieser Komplex hält das Zytokin in einem inaktiven Zustand, in dem TGFβ nicht an Rezeptor binden kann. Die Prodomäne von TGFβ, latency-associated peptide (LAP) genannt, und TGFβ bilden mit einem weiteren LAP/TGFβ Komplex einen Homodimer. Die beiden LAP Proteine formen dadurch eine Klammer um den TGFβ Dimer herum. Andere Vertreter der TGFβ-Familie können auch Heterodimere formen[9]. Ob dies auch bei TGFβ1-3 möglich ist, ob also z. B. ein TGFβ1 mit einem TGFβ2 einen Dimer formen kann, ist derzeit unklar.
Die "LAP-Klammer" um den TGFβ1-Dimer wird durch eine Disulfidbrücke an einem Ende von LAP stabilisiert. TGFβ selbst wird im sogenannten Cystin-Knoten durch vier Disulfidbrücken intramolekular stabilisiert; eine weitere Disulfidbrücke bildet intermolekular die Verbindung zum zweiten TGFβ und formt dadurch den Homodimer[1]. Dieser Cystin-Knoten ist nicht nur typisch für die gesamte TGFβ-Familie, sondern auch für die übergeordnete Superfamilie der Cystin-Knoten Wachstumsfaktoren (cystein knot growth factor, CKGF superfamily)[9]. Zu dieser CKGF-Superfamilie gehört außer der TGFβ-Familie auch die Dan-Familie (BMP Antagonisten), die Glykoprotein Hormonfamilie (z. B. FSH), die Familie der Bursicon Hormone (nur in Wirbellosen zu finden), die platelet-derived growth factor (PDGF) Familie, und die Familie der nerve growth factors (NGFs)[9].
Die TGFβ Zytokine haben eine sehr stark konservierte Aminosäuresequenz. Proteinstrukturen für alle drei TGFβ Zytokine sind verfügbar und zeigen auch eine hohe strukturelle Ähnlichkeit. Für komplette LAP-TGFβ sind dagegen nur für TGFβ1 Proteinstrukturen verfügbar. Da die verschiedenen LAP Proteine wesentlich weniger gut evolutionär konserviert sind[9], ist zu erwarten, dass sich die verschiedenen TGFβ in diesem Bereich auch strukturell mehr unterscheiden.
Der Komplex von LAP und TGFβ kann von Zellen sekretiert werden. Häufig ist der LAP-TGFβ Komplex aber mittels LAP an ein weiteres Protein gebunden, bevor der Komplex sekretiert wird. In den meisten Zellen ist dies eine Bindung von LAP an das Protein LTBP (latent TGFβ binding protein). In regulatorischen T-Zellen und Thrombozyten wird dagegen das Protein GARP exprimiert und der LAP-TGFβ Komplex bindet an GARP. Ein LAP/TGFβ/LTBP-Komplex ist über LTBP an Proteine der EZM gebunden; häufig Fibronektin oder Fibrillin[10]. Ein LAP/TGFβ/GARP-Komplex wird dagegen über GARP an der Zelloberfläche präsentiert. Beide LAP Proteine des Homodimers binden jeweils mit Disulfidbrücken an LTBP, bzw. GARP und stabilisieren damit ebenfalls den LAP/TGFβ-Homodimer.
Aktivierung
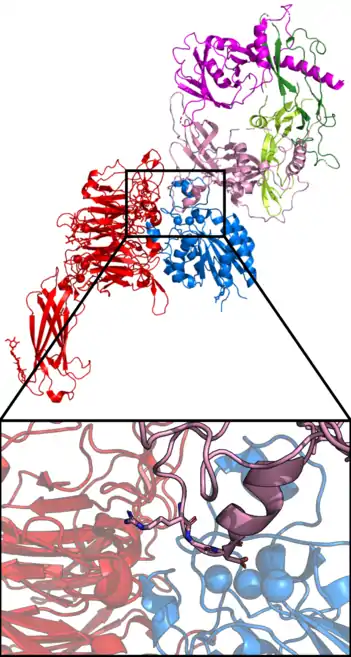
Zu Beginn der Entdeckung von TGFβ war unklar, dass es nur inaktiv von Zellen sekretiert wird. Die biochemischen Protokolle zur Reinigung und Konzentration von TGFβ enthielten Säuren, die in der Lage sind TGFβ aus dem Komplex mit LAP zu lösen[11]. Die Bedeutung der Aktivierung von TGFβ wurde daher zu Beginn nicht erkannt, da man nur mit TGFβ arbeitete, das unwissentlich bereits aktiviert wurde.
Im Laufe der Jahre wurden mehr und mehr Prozesse bekannt, die prinzipiell in der Lage sind TGFβ zu aktivieren. Ausgehend von verschiedenen Studien mit Knockout-Mäusen scheint es derzeit, dass bestimmte Integrine für die Aktivierung von TGFβ1 und TGFβ3 am wichtigsten sind[10]. Allerdings wurde kein Integrin gefunden, das an TGFβ2 bindet. Dies bedeutet entweder, dass TGFβ2 ausschließlich Integrin-unabhängig aktiviert wird, oder dass eine TGFβ2-Integrin Bindung bisher noch nicht entdeckt wurde. Auch für TGFβ1 und TGFβ3 besteht weiterhin die Möglichkeit, dass Integrin-unabhängige Aktivierungsmechanismen zumindest in bestimmten Situationen dominieren.
Integrin-abhängig
Sowohl TGFβ1, als auch TGFβ3 haben in ihrem LAP Protein eine RGD-Peptidsequenz. Diese Aminosäuresequenz wird von vielen Integrinen erkannt. Die Bedeutung der RGD-Integrin-Bindung für die Aktivierung von TGFβ1 zeigt sich bei Knockin-Mäusen, bei denen TGFβ1-RGD durch RGE ersetzt wird (die RGE-Sequenz reduziert die Integrin-Bindung oder verhindert sie komplett). Diese TGFβ1-RGE Mäuse zeigen ähnlich Effekte wie Mäuse, denen TGFβ1 komplett fehlt[12]. D. h., dass eine fehlende Aktivierung von TGFβ1 durch ein Integrin gleichbedeutend ist mit der kompletten Abwesenheit von TGFβ1.
Zwei Integrine, αVβ6 und αVβ8 Integrin, werden derzeit als wichtigste Integrine für die Aktivierung von TGFβ1 und TGFβ3 in Organismen diskutiert[13]. Neuere Publikationen deuten aber auch eine große Bedeutung von αVβ1 Integrin an[14]. Zumindest in Zellkultur haben außerdem auch αVβ3, αVβ5 und α8β1 Integrine eine Bindung an TGFβ1 gezeigt. Strukturelle Erwägungen legen bisher aber nahe, dass zumindest αVβ3 und αVβ5 Integrin nicht ohne weiteres an RGD in TGFβ1 binden können[15] und damit eher in Ausnahmesituationen TGFβ1 aktivieren könnten.
Während sowohl αVβ6 als auch αVβ8 Integrin an die RGD-Sequenz in TGFβ1 und TGFβ3 binden, ist der jeweilige Aktivierungsmechanismus durch die beiden Integrine verschieden.
Aktivierung mittels αVβ6 Integrin
Die Aktivierung von TGFβ mittels Integrin beruht auf konformationeller Änderung im LAP Protein durch Zugkraft. Damit dies möglich ist, muss der LAP/TGFβ-Homodimer an einem Ende stabil verankert sein, während am anderen Ende Zellen mittels αVβ6 Integrin an einem LAP-Protein ziehen müssen. Eine stabile Verankerung erfolgt durch eine Verknüpfung an EZM-Proteine mittels LTBP oder an die Oberfläche einer benachbarten Zelle durch GARP. Die Zugkraft durch die Zelle ist bei Bindung durch αVβ6 Integrin gegeben, da dieses Integrin mittels fokaler Adhäsionen an kontraktile Aktinfilamente verknüpft ist. Molekulare Simulationen basierend auf einer αVβ6-LAP/TGFβ1-Struktur zeigen wie mechanischer Zug zu konformationellen Änderungen im LAP-Bereich führt, die schließlich TGFβ1 aus der LAP-Klammer befreien[16]. Experimentell kann man nachweisen, dass freies, aktives TGFβ1 durch diesen Prozess von der Bindung an EZM-Proteine frei wird und löslich im Medium vorliegt.
Andere Integrine wie αVβ1, αVβ3, αVβ5 und α8β1 sind wie αVβ6 in der Lage durch Aktinfilamente Zugkräfte in die EZM zu übertragen. Falls diese Integrine eine Rolle für die TGFβ-Aktivierung in Organismen spielen, werden sie vermutlich ebenfalls diesen hier beschriebenen Mechanismus zur TGFβ-Aktivierung benutzen.
Aktivierung mittels αVβ8 Integrin
β8 Integrin hat eine cytoplasmatische Aminosäurensequenz, die sich stark von der aller anderen β Integrine unterscheidet. Dadurch ist αVβ8 Integrin nicht in der Lage fokale Adhäsionen auszubilden und sich über Adapterproteine an kontraktile Aktinfilamente zu binden. Eine Aktivierung von TGFβ über Zugkraft kommt daher für αVβ8 Integrin sehr wahrscheinlich nicht in Frage. Es wurde bisher vermutet, dass die Bindung von TGFβ an αVβ8 Integrin eher die Rekrutierung von Proteasen ermöglicht, welche dann die LAP-Klammer öffnen, damit TGFβ freigesetzt werden kann und zu TGFβ-Rezeptoren diffundieren kann[10]. Mittlerweile hat sich aber gezeigt, dass αVβ8 Integrin eine Aktivierung von TGFβ1 ermöglicht, bei der TGFβ1 nicht frei gesetzt wird, aber trotzdem in der Lage ist an TGFβ-Rezeptoren zu binden[17].
Integrin-unabhängig
Verschiedene Integrin-unabhängige Aktivierungsmechanismen von TGFβ werden ebenfalls diskutiert[18]:
- Plasmin
- Proteasen (u. a. MMP-9, MMP-13)
- Thrombospondin
- Scherkräfte
- Sauerstoffradikale (ROS, reactive oxygen species)
- pH-Wert
Im Vergleich zur Aktivierung durch Integrine sind die Belege für die TGFβ-Aktivierung durch die hier genannten Faktoren weniger konsistent[10]. Dies wird auch durch ein evolutionsbiologisches Argument unterstützt: Integrine waren bereits vorhanden als TGFβ entstanden. Plasmin, die genannten Proteasen und Thrombospondin entstanden dagegen erst später. Dies bedeutet jedoch nicht, dass Integrin-unabhängige Aktivierungsmechanismen in bestimmten Situationen nicht über TGFβ-Aktivierung entscheiden könnten.
Signalwege
Der TGFβ-Signalweg besteht aus der Bindung des Wachstumsfaktors an den Rezeptorkomplex an der Zelloberfläche. Dieser aktiviert intrazellulär Signalmoleküle der Smad-Familie, welche wiederum die Genregulation beeinflussen.
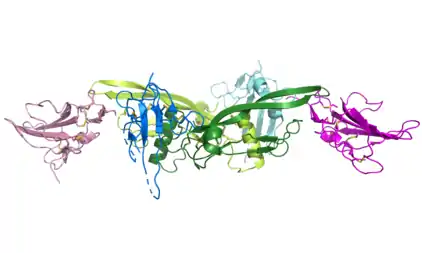
Rezeptoren
Der kanonische TGFβ Signalweg erfolgt durch Aktivierung von TGFβ-Rezeptoren, die zu den Serin/Threonin-Kinasen zählen. Dafür bindet ein TGFβ-Homodimer an zwei TGFβ-Rezeptor Typ-II (TbR-II). Dieser Präkomplex rekrutiert dann wiederum zwei TGFβ-Rezeptor Typ-I (TbR-I)[8]. TGFβ1 und TGFβ3 haben eine höhere Affinität für TbR-II als für TbR-I, was die Abfolge der Bindung von TGFβ an die Rezeptor-Untereinheiten erklärt. Im Gegensatz dazu hat TGFβ2 etwa gleiche Affinitäten für TbR-I und TbR-II. Zusätzliche Korezeptoren (Betaglykan) können aber dafür sorgen, dass TGFβ2 in ähnlicher Abfolge erst TbR-II und dann TbR-I bindet[9].
Generell ist die Affinität zwischen TGFβ und dem TbR-I / TbR-II - Rezeptorkomplex höher als zwischen Rezeptor-Tyrosinkinasen und den daran bindenden Wachstumsfaktoren wie EGF. Gleichzeitig ist die Zahl von TbR-I und TbR-II an der Zelloberfläche mit 5 000 oder weniger sehr gering im Vergleich zu Rezeptor-Tyrosinkinasen[8]. Dies ermöglicht es Zellen potenziell sehr dynamisch ihre TGFβ-Signalkapazitäten anzupassen, da Hinzufügen oder Entfernen von vergleichsweise wenigen Rezeptoren an der Zelloberfläche prozentual bereits einen großen Unterschied macht.
Smad-regulierte Genexpression
Nach der Rekrutierung von TbR-I durch den TGFβ-TbR-II Komplex wird TbR-I durch TbR-II phosphoryliert und damit aktiviert. In diesem Zustand ist TbR-I in der Lage Rezeptor-regulierte Smad Proteine (R-Smads: Smad2, 3, 5, 8) zu rekrutieren und zu aktivieren[8]. Ein Komplex von zwei dieser Smads formt mit Smad4 einen Komplex, der im Zellkern an DNA bindet und mit anderen Transkriptionsfaktoren die Genexpression beeinflusst. Andere Signalwege können diesen Prozess auf mehreren Ebenen beeinflussen. Diese Regulation durch andere Signalwege, sowie die kontextabhängigen Kofaktoren der Genexpression, machen es schwierig die beeinflussten Gene des TGFβ-Smad-Signalwegs exakt vorherzusagen[8]. Im Rahmen der oben beschriebenen Wirkung von TGFβ auf EMT und Wundheilung gehören aber Rekrutierung von Transkriptionsfaktoren wie Snail (regulieren EMT) und erhöhte Produktion und Sekretion mehrerer EZM-Proteine wie Kollagene zu den typischen Effekten des TGFβ-Smad-Signalwegs.
Klinische Relevanz
Die Inhibierung des TGFβ-Signalwegs erfährt in den letzten Jahren ein vermehrtes klinisches Interesse. Dies liegt zum einen am immundämpfenden Effekt von TGFβ. Tumore können zu einer vermehrten Aktivierung von TGFβ führen und sich durch die Wirkung von TGFβ vor einer Attacke durch das Immunsystem schützen.
Zum anderen führt andauernde TGFβ-Aktivierung wie es bei Entzündungen oder Wunden erfolgt zu einer Fibrose, die bis zum Organversagen führen kann. Es wird geschätzt, dass in entwickelten Ländern mehr als 40 % aller Todesfälle mit einer fibrotischen Erkrankung in Verbindung stehen.
Das Engelmann-Syndrom wird durch eine Genmutation in Chromosom 19 Genlocus q13.1–13.3 verursacht. Betroffen ist die β-1-Kette des Transforming growth factor (TGF β1).[19]
Es wird vermutet, dass TGF-β eine Schlüsselrolle bei der Pathogenese der strahlenbedingten Lungenfibrose einnimmt, eventuell lässt sich durch Antagonisierung von TGF-β eine solche Entzündung verhindern. In dieser Frage liegen aber bislang wenige bis keine Forschungsergebnisse vor.
TGF-β scheint auch bei der Entstehung der diabetischen Nierenschädigung beteiligt zu sein. Bei der diabetischen Nephropathie kommt es zu einer Zellvergrößerung (Hypertrophie) und zu einer vermehrten Bildung von Kollagen im Nierenkörperchen. Ein erhöhter Blutzucker stimuliert die Freisetzung von TGF-β. Wird TGF-β durch ACE-Hemmer, spezifische Antikörper oder Hepatocyte Growth Factor gehemmt, führt dies zu einer Besserung der diabetischen Nierenschädigung.
Bei Patienten mit vaskulärem Typ des Ehlers-Danlos-Syndroms und bei Patienten mit Loeys-Dietz-Syndrom wurden Mutationen in den Genen des Rezeptors für TGF-β (TGFBR1 und TGFBR2) nachgewiesen.[20]
Das Marfan-Syndrom wird durch eine Mutation im Gen für Fibrillin hervorgerufen. Fibrillin ist ein Bestandteil der Mikrofibrillen des Bindegewebes. So führt die Mutation zu einer verminderten Festigkeit des Bindegewebes. Fibrillin ist aber auch homolog zur Familie der Latenten TGF-β bindenden Proteine (LTBP), die TGF-β in einen inaktiven Komplex binden. Die Mutation im Gen des Fibrillin führt zu einer verminderten Bindung von TGF-β. Der resultierende Überschuss von aktivem TGF-β im Bindegewebe wird für einige Komplikationen des Marfan-Syndroms verantwortlich gemacht, wie Lungenemphysem, Mitralklappenprolaps und Aneurysmen der Aortenwurzel.[21]
Ciclosporin ist ein wichtiges Medikament in der Transplantationsmedizin. Gravierende Nebenwirkungen sind Bluthochdruck und Nierenschäden. Eine mögliche Ursache dieser Komplikationen ist, dass die Transkription von TGF-β durch Ciclosporin stimuliert wird.[22]
Siehe auch
Einzelnachweise
- Masato Morikawa, Rik Derynck, Kohei Miyazono: TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. In: Cold Spring Harbor Perspectives in Biology. Band 8, Nr. 5, Mai 2016, ISSN 1943-0264, S. a021873, doi:10.1101/cshperspect.a021873, PMID 27141051, PMC 4852809 (freier Volltext).
- Marcia M. Shull, Ilona Ormsby, Ann B. Kier, Sharon Pawlowski, Ronald J. Diebold: Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. In: Nature. Band 359, Nr. 6397, Oktober 1992, ISSN 0028-0836, S. 693–699, doi:10.1038/359693a0, PMID 1436033, PMC 3889166 (freier Volltext).
- L. P. Sanford, I. Ormsby, A. C. Gittenberger-de Groot, H. Sariola, R. Friedman: TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. In: Development (Cambridge, England). Band 124, Nr. 13, Juli 1997, ISSN 0950-1991, S. 2659–2670, PMID 9217007, PMC 3850286 (freier Volltext) – (Online [abgerufen am 1. Mai 2020]).
- G. Proetzel, S. A. Pawlowski, M. V. Wiles, M. Yin, G. P. Boivin: Transforming growth factor-beta 3 is required for secondary palate fusion. In: Nature Genetics. Band 11, Nr. 4, Dezember 1995, ISSN 1061-4036, S. 409–414, doi:10.1038/ng1295-409, PMID 7493021, PMC 3855390 (freier Volltext).
- Vesa Kaartinen, Jan Willem Voncken, Charles Shuler, David Warburton, Ding Bu: Abnormal lung development and cleft palate in mice lacking TGF–β3 indicates defects of epithelial–mesenchymal interaction. In: Nature Genetics. Band 11, Nr. 4, Dezember 1995, ISSN 1061-4036, S. 415–421, doi:10.1038/ng1295-415.
- Boris Hinz: Myofibroblasts. In: Experimental Eye Research. Band 142, Januar 2016, S. 56–70, doi:10.1016/j.exer.2015.07.009 (elsevier.com [abgerufen am 1. Mai 2020]).
- Boris Hinz: The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. In: Matrix Biology. Band 47, September 2015, S. 54–65, doi:10.1016/j.matbio.2015.05.006 (elsevier.com [abgerufen am 1. Mai 2020]).
- Rik Derynck, Erine H. Budi: Specificity, versatility, and control of TGF-β family signaling. In: Science Signaling. Band 12, Nr. 570, 26. Februar 2019, ISSN 1945-0877, S. eaav5183, doi:10.1126/scisignal.aav5183, PMID 30808818, PMC 6800142 (freier Volltext).
- Andrew P. Hinck, Thomas D. Mueller, Timothy A. Springer: Structural Biology and Evolution of the TGF-β Family. In: Cold Spring Harbor Perspectives in Biology. Band 8, Nr. 12, Dezember 2016, ISSN 1943-0264, S. a022103, doi:10.1101/cshperspect.a022103, PMID 27638177, PMC 5131774 (freier Volltext).
- Ian B. Robertson, Daniel B. Rifkin: Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. In: Cold Spring Harbor Perspectives in Biology. Band 8, Nr. 6, Juni 2016, ISSN 1943-0264, S. a021907, doi:10.1101/cshperspect.a021907, PMID 27252363, PMC 4888822 (freier Volltext).
- Harold L. Moses, Anita B. Roberts, Rik Derynck: The Discovery and Early Days of TGF-β: A Historical Perspective. In: Cold Spring Harbor Perspectives in Biology. Band 8, Nr. 7, Juli 2016, ISSN 1943-0264, S. a021865, doi:10.1101/cshperspect.a021865, PMID 27328871, PMC 4930926 (freier Volltext).
- Zhiwei Yang, Zhenyu Mu, Branka Dabovic, Vladimir Jurukovski, Dawen Yu: Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. In: Journal of Cell Biology. Band 176, Nr. 6, 12. März 2007, ISSN 1540-8140, S. 787–793, doi:10.1083/jcb.200611044, PMID 17353357, PMC 2064053 (freier Volltext) – (rupress.org).
- P. Aluwihare, Z. Mu, Z. Zhao, D. Yu, P. H. Weinreb: Mice that lack activity of v 6- and v 8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. In: Journal of Cell Science. Band 122, Nr. 2, 15. Januar 2009, ISSN 0021-9533, S. 227–232, doi:10.1242/jcs.035246, PMID 19118215, PMC 2714418 (freier Volltext).
- Nilgun I. Reed, Hyunil Jo, Chun Chen, Kazuyuki Tsujino, Thomas D. Arnold: The α v β 1 integrin plays a critical in vivo role in tissue fibrosis. In: Science Translational Medicine. Band 7, Nr. 288, 20. Mai 2015, ISSN 1946-6234, S. 288ra79–288ra79, doi:10.1126/scitranslmed.aaa5094, PMID 25995225, PMC 4461057 (freier Volltext).
- Michael Bachmann, Sampo Kukkurainen, Vesa P. Hytönen, Bernhard Wehrle-Haller: Cell Adhesion by Integrins. In: Physiological Reviews. Band 99, Nr. 4, 1. Oktober 2019, ISSN 0031-9333, S. 1655–1699, doi:10.1152/physrev.00036.2018.
- Xianchi Dong, Bo Zhao, Roxana E. Iacob, Jianghai Zhu, Adem C. Koksal: Force interacts with macromolecular structure in activation of TGF-β. In: Nature. Band 542, Nr. 7639, Februar 2017, ISSN 0028-0836, S. 55–59, doi:10.1038/nature21035, PMID 28117447, PMC 5586147 (freier Volltext).
- Melody G. Campbell, Anthony Cormier, Saburo Ito, Robert I. Seed, Andrew J. Bondesson: Cryo-EM Reveals Integrin-Mediated TGF-β Activation without Release from Latent TGF-β. In: Cell. Band 180, Nr. 3, Februar 2020, S. 490–501.e16, doi:10.1016/j.cell.2019.12.030 (elsevier.com [abgerufen am 1. Mai 2020]).
- J. S. Munger, D. Sheppard: Cross Talk among TGF - Signaling Pathways, Integrins, and the Extracellular Matrix. In: Cold Spring Harbor Perspectives in Biology. Band 3, Nr. 11, 1. November 2011, ISSN 1943-0264, S. a005017–a005017, doi:10.1101/cshperspect.a005017, PMID 21900405, PMC 3220354 (freier Volltext).
- Camurati-Engelmann disease – Genetics Home Reference. In: ghr.nlm.nih.gov. 23. Februar 2015, abgerufen am 25. Februar 2015.
- Bart L. Loeys u. a.: Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. In: New England Journal of Medicine. Nr. 355, 2006, S. 788–798 (Abstract).
- Bruce D. Gelb: Marfan's Syndrome and Related Disorders — More Tightly Connected Than We Thought. In: New England Journal of Medicine. Nr. 355, 2006, S. 841–844 (Abstract).
- Prashar Y: Stimulation of transforming growth factor-beta 1 transcription by cyclosporine. In: FEBS Lett. Nr. 358, 1995, S. 109–112, PMID 7828718.