Ehlers-Danlos-Syndrom
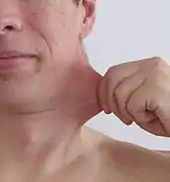
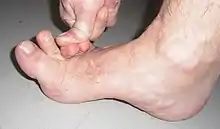
Das Ehlers-Danlos-Syndrom (EDS) zählt zu den seltenen Krankheiten. Genauer gesagt, ist EDS ein Sammelbegriff für eine heterogene Gruppe von seltenen angeborenen Störungen im Bindegewebe. Die wichtigsten sichtbaren gemeinsamen Erkennungsmerkmale dieser Gruppe sind eine Überdehnbarkeit der Haut und überbewegliche Gelenke. Betroffen sind jedoch auch Gefäße, Muskeln, Bänder, Sehnen und innere Organe. Die Erkrankung kann durch das schwache Bindegewebe tödlich verlaufen, beispielsweise durch spontane Organrupturen, Aortendissektion oder Aneurysmen.
| Klassifikation nach ICD-10 | |
|---|---|
| Q79.6 | Ehlers-Danlos-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Die Erkrankung verläuft in den meisten Fällen fortschreitend.
Bisher sind neunzehn Genmutationen bekannt, die EDS auslösen.[1] Die verschiedenen Mutationen führen zu einer Veränderung der Struktur, der Produktion oder der Verarbeitung von Kollagen oder von Proteinen, die mit Kollagen interagieren.[1] Die Häufigkeit des Auftretens in der Bevölkerung wird mit 1:20.000 angenommen, somit handelt es sich bei EDS um eine seltene Erkrankung. Der häufigste Typ ist der hypermobile Typ mit einer Prävelenz von 1:5.000. Die seltensten Typen treten nur wenige Male weltweit auf.[2] Es gibt keine Unterschiede im Auftreten zwischen verschiedenen ethnischen Gruppen oder den Geschlechtern.

Geschichte
Das Syndrom ist eine der ältesten bekannten Ursachen von Ergüssen und Blutungen, die schon von Hippokrates 400 v. Chr. erkannt wurden.[3] Eine erste Fallanalyse mit abnormer Hautelastizität eines spanischen Mannes ist durch den niederländischen Chirurgen Job Janzoon von Meerkerin aus dem Jahre 1668 bekannt.[4] Die erste umfassende Beschreibung des Syndroms mit seinen vielen Facetten (Haut, Gelenke, Narben usw.) entstand 1891 durch den russischen Dermatologen Tschernogubow.[5] Das isolierte Zarenreich verhinderte jedoch das allgemeine Bekanntwerden der Studie, sodass Edvard Ehlers mit der Beschreibung der wesentlichen Zusammenhänge 1901 als Cutis laxa[6] und Henri-Alexandre Danlos 1908 mit dem Vorschlag, die Überdehnbarkeit und Zerreißbarkeit der Haut als Kardinalsymptome zu benennen[7], die Namensgeber des Syndroms wurden.
Andere vorwiegend veraltete Bezeichnungen des EDS waren:
- Ehlers-Danlos-Meekeren-Syndrom, Meekeren-Ehlers-Danlos-Syndrom, Van-Meekeren-Syndrom
- Danlos-Syndrom
- Fibrodysplasia elastica generalisata, Cutis hyperelastica
- Dermatolyse
- Gummihaut, Indian rubber skin
- Tschernogubow-Syndrom
- Sack-Barabas-Syndrom
Klassifikation
Eine EDS-Klassifikation wurde ab den späten 1960er Jahren versucht. 1986 definierte man dann zehn Typen, die 1988 anlässlich einer Konferenz in Berlin veröffentlicht wurden. Mit fortschreitenden Erkenntnissen auf molekularem und biochemischem Gebiet wurde 1997 EDS neu unterteilt. Die Villefranche-Klassifikation diente einer klinisch-vereinfachten Diagnostik des Ehlers-Danlos-Syndroms und zur Abgrenzung von Erkrankungen, die mit dem EDS überlappen (siehe Tabelle).[8] Es ließ sich nun die Krankheit durch sechs Typen mit ihren Haupt- und Nebenkriterien beschreiben. Darüber wurden weitere andere, exotische Formen von EDS in Einzelfällen identifiziert. Seit 2017 wird die Villefranche-Klassifikation durch die neuen Diagnosekriterien abgelöst.[9] Die Kriterien wurden vollständig überarbeitet und aktualisiert und unterscheiden nun in 13 Typen. Folgende Beschwerdebilder werden nach den neuen Diagnosekriterien nicht mehr dem EDS Spektrum zugeordnet: Okzipitalhorn-Syndrom, Fibronectin-Mangel (EDS Typ X), familiäres Hypermobilitätssyndrom (EDS Typ XI), X-linked EDS (EDS Typ V) und Filamin A EDS.[10]
Es besteht eine große Variabilität und Überlappung in den Symptomen zwischen den einzelnen Typen. Eine eindeutige Klassifizierung ist somit nur anhand klinischer Diagnostik und genetischer Tests bzw. Hautbiopsien möglich.
Die auslösenden Mutationen wurden identifiziert.[10] Einzig beim hypermobilen Typ ist das auslösende Gen noch nicht bekannt.[11]
Neue Diagnosekriterien 2017
Zeitgleich mit der Veröffentlichung von 18 Artikeln zu den Subtypen des Ehlers-Danlos-Syndroms im American Journal of Medical Genetics wurden im März 2017 auch neue Diagnosekriterien veröffentlicht. Diese sind weitaus umfassender und spezifischer als die vorherigen Villefranche-Kriterien. Ein internationales Komitee hat in verschiedenen Arbeitsgruppen die Erfahrungen der klinischen Untersuchungen und die Ergebnisse genetischer Studien zusammengeführt, auch um die Sensibilität der Ärzte für dieses Krankheitsbild zu verbessern und durch länderübergreifende Kriterien die Diagnostik und somit die Versorgung der Patienten zu verbessern. Dafür sollen neben den konkreteren Möglichkeiten zur Diagnostik auch Richtlinien zur Behandlung erarbeitet werden.
Die neuen Diagnosekriterien lösen die Villefranche-Klassifikation ab. Das Ehlers-Danlos-Syndrom wird nun in 13 Typen unterschieden.
Hypermobiles EDS (hEDS, früher Typ 3): Die genetische Ursache ist bisher nicht bekannt. Die Diagnose wird nach eingehender Untersuchung klinisch gestellt.[12] hEDS zeichnet sich vor allem durch eine Hypermobilität großer und kleiner Gelenke aus. Subluxationen und Luxationen können regelmäßig vorkommen. hEDS Betroffene leiden oft unter Gelenkinstabilitäten und weisen häufig eine weiche, samtige Haut auf, die leicht verletzlich sein kann.[13] Sehr häufig leiden Patienten unter chronischen Schmerzen. Die Ausprägung kann mäßig bis sehr stark sein. Rund 90 % der EDS Betroffenen weisen chronische Schmerzen auf, wobei die höchsten Schmerz-Scores bei hEDS Patienten gefunden wurden.[14]
Klassisches EDS (cEDS, früher Typ 1 und 2): Die genetische Ursache für diesen EDS Typ findet sich zu rund 90 % in den Genen COL5A2 und COL5A. Selten liegen Mutationen in COL1A1 vor.[15] Major-Kriterien sind eine extrem elastische Haut, die fragil und leicht verletzlich ist, atrophische Narbenbildung und eine allgemeine Gelenkhypermobilität. Daneben gibt es neun "Minor"-Kriterien, wie samtige Haut, Muskelhypotonie oder Pseudotumore.[16]
Vaskuläres EDS (vEDS, früher Typ 4): Die genetische Ursache lässt sich in Variationen des COL3A1 Gens lokalisieren. Es gibt fünf Major-Kriterien und 12 Minor-Kriterien, die einen klinischen Anhalt auf vEDS geben. Patienten weisen häufig eine dünne, durchscheinende Haut auf, die sehr fragil und leicht verletzlich ist. Adern und Organe sind ebenso leicht verletzlich. Die Gelenke sind überbeweglich; meist handelt es sich dabei um kleine Gelenke wie Finger oder Zehen. Klumpfüße oder Bänder- und Muskelrisse können auftreten. vEDS ist lebensbedrohlich; im Median werden Betroffene 51 Jahre alt. Die Lebensspanne ist dabei weit und reicht von rund zehn bis ungefähr 80 Jahre. Häufigste Todesursache sind arterielle Dissektionen oder Rupturen.[17]
Kyphoskoliotisches EDS (kEDS, früher Typ 6): Auslösend sind Varianten im Gen PLOD1, seltener im FKBP 14 Gen. Major-Kriterien sind angeborene Muskelhypertonie, angeborene oder früh einsetzende Kyphoskoliose und eine generalisierte Hypermobilität mit (Sub-)Luxationen. Hinzu kommen Minor-Kriterien. Die Kyphoskoliose ist meist schwerwiegend und progressiv. Weitere Symptome können eine weiche, teigige Haut, atrophische Narbenbildung und schlechte Wundheilung umfassen.[15]
Arthrochalasie EDS (aEDS, früher Typ 7 A und B): aEDS wird durch Mutationen in den Genen COL1A1 und COL1A2 verursacht. Derzeit sind rund 30 Fälle bekannt. Betroffene leiden unter ausgeprägter Gelenkhypermobilität und angeborener, bilateraler Hüftluxation. Hinzukommen (Sub-)Luxationen der großen und kleinen Gelenke, Fußdeformitäten und Skoliose, Lordose und Kyphoskoliose können auftreten. Die Haut kann überdehnbar, samtig und leicht verletzlich sein.[15]
Dermatosparaxis EDS (früher Typ 7C): Mutationen im ADAMTS2-Gen lösen dEDS aus. Symptome umfassen eine extrem fragile Haut, die schnell Narben bildet. Die Haut ist lose und überschüssig vorhanden. Die Gelenküberbeweglichkeit reicht von mild ausgeprägt hin zu schwerwiegend. dEDS ist sehr selten – bisher sind rund zehn Fälle bekannt.[15]
Brittle cornea Syndrom (BCS): BCS tritt in zwei Typen auf. Variante 1 entsteht durch eine Mutation im ZNF469 Gen, Variante 2 durch eine Mutation im PRDM5 Gen. BCS ist durch eine voranschreitende Verdünnung der Hornhaut des Auges charakterisiert. Früh können ein Keratoglobus oder Keratokonus auftreten, sowie Kurzsichtigkeit, Hörverlust und blaue Skleren. Klassische EDS Symptome wie überbewegliche Gelenke und überdehnbare Haut werden häufig beobachtet.[15]
Classical-like EDS (clEDS): Mutationen im TNXB Gen führen zu clEDS. Die Haut ist meist überdehnbar und samtig, weist aber keine atrophische Narbenbildung auf. Häufig liegt eine allgemeine Gelenkshypermobilität vor, (Sub-)Luxationen können auftreten, vor allem sind die Schultern und Fußgelenke betroffen.[15] Die Prävelenz liegt laut OrphaNet bei weniger als 1:1.000.000.
Spondylodysplastisches EDS (spEDS): spEDS wird ausgelöst durch Mutationen in den Genen B4GALT7, B3GALT6 und SLC39A13. Betroffene sind häufig klein und weisen eine Muskelhypotonie auf.[15]
Muskulocontrakturelles EDS (mcEDS): Auslösend für mcEDS sind Mutationen in den Genen CHST14 und DSE. Charakteristisch ist eine Hautüberdehnbarkeit, eine erhöhte Hautverletzlichkeit, atrophische Narbenbildung und eine erhöhte Faltenbildung in den Handflächen. mcEDS führt zu angeborenen Kontrakturen.[15]
Myopathisches EDS (mEDS): Mutationen im COL12A1 Gen führen zu mEDS. mEDS-Betroffene leiden an einer angeborenen Muskelhypotonie und/oder an Muskelatrophie, die sich mit zunehmendem Alter bessert. Proximale Gelenkkontrakturen und eine Gelenkhypermobilität distaler Gelenke können auftreten.[15]
Periodontales EDS (pEDS): pEDS wird durch Mutationen im C1R Gen ausgelöst. pEDS führt zu schwerwiegender, hartnäckiger Parodontitis, die bereits in der Kindheit oder im Jugendalter auftritt. Das Zahnfleisch liegt oft nicht ausreichend am Zahn an.[15]
Kardio-valvuläres EDS (cvEDS): COL1A2- können zu cvEDS führen. Schwerwiegend fortschreitende kardio-valvuläre Symptome, wie an der Mitralklappe, und eine überdehnbare, leicht verletzliche Haut, die zu atrophischer Narbenbildung neigt, charakterisieren cvEDS. Außerdem tritt eine Gelenkhypermobilität auf, die entweder generalisiert oder auf die kleinen Gelenke begrenzt sein kann.[15]
Die EDS-Typen können auch nach Gruppen sortiert werden. Die Einordnung erfolgt in diesem Fall nach Gemeinsamkeiten der auslösenden Gene.[18]
Gruppe A: Störungen der Kollagenprimärstruktur und der Kollagenverarbeitung, dies umfasst cEDS, vEDS, aEDS, dEDS und cvEDS.
Gruppe B: Störungen der Kollagenfaltung und des Kollagen crosslinking, dies umfasst kEDS-PLOD1 und kEDS-FKB14.
Gruppe C: Störungen der Struktur und der Funktion der Myomatrix, dies umfasst clEDS und mEDS.
Gruppe D: Störungen der glycosaminoglycan Biosynthese, dies umfasst spEDS-B4GALT7, spEDS-b3GALT6, mcEDS-CHST14 und mcEDS-DSE.
Gruppe E: Defekte im Komplementsystem, dies umfasst pEDS.
Gruppe F: Störungen von intrazellulären Prozessen, dies umfasst spEDS-SLC39A13 und BCS.
Gruppe G: Derzeit ungeklärt, dies umfasst hEDS.
| Frühere Klassifikation nach „Villefranche“ | Frühere „Berlin“-Klassifikation | bekannte Gene ("Villefranche" Klassifikation) | Charakteristika |
|---|---|---|---|
| Klassischer Typ | EDS Typ I (gravis) und Typ II (mitis) | COL5A1/COL5A2- und COL1A1-Mutationen
|
stark überdehnbare und leicht verletzbare Haut; Hämatomneigung; abnorme Wundheilung; Überbeweglichkeit der Gelenke; innere Organe und Gefäße mit betroffen (Symptome bei Typ II wie bei Typ I, nur geringer ausgeprägt)[23] (Autosomal-dominanter Erbgang)[24] Gorlin-Zeichen, mit der Zunge kann die Nase berührt werden.[25] |
| Hypermobiler Typ | Hypermobil – EDS Typ III[26] | Keine Gene bekannt.[27][28][29][30][31] | nur leicht überdehnbare, seidige Haut; ausgeprägte Überbeweglichkeit der Gelenke; häufige (Sub-)Luxationen der Gelenke, Sehnenentzündungen, Bandscheibenvorfälle etc.; chronische Glieder- und Gelenkschmerzen, Familienhistorie ("The diagnosis of hEDS is based entirely on clinical evaluation and family history.")[32] Umgekehrtes-Namaskar-Zeichen, Innenhandflächen können hinter dem Rücken gegeneinander gedrückt werden.[33][34] |
| Vaskulärer Typ | Arterielle Form – EDS Typ IV | COL3A1[35] | dünne, durchscheinende Haut; ausgeprägte Hämatomneigung; Überbeweglichkeit der kleinen Gelenke; Beteiligung der inneren Organe und Gefäße; spontane Ruptur von Arterien und Gefäßen, Aneurysmen (Autosomal-dominante Vererbung)[36][37] |
| Kyphoskoliose Typ | Okular-Skoliose – EDS Typ VI | PLOD1 | Überdehnbarkeit der Haut mittel bis stark; abnorme Wundheilung; starke Überbeweglichkeit der Gelenke; Augenbeteiligung; Beteiligung der inneren Organe, veraltete Bezeichnung: Nevo-Syndrom[38][39][40] (Autosomal-rezessive Vererbung)[24] |
| Arthrochalasie Typ | Arthrochalasie multiplex – EDS Typen VIIA und VIIB | COL1A1/COL1A2, teilweises o. komplettes Fehlen von exon 6 | Überdehnbarkeit der Haut (gering bis mittel), dünne Haut, Hüftluxation, ausgeprägte Überbeweglichkeit der Gelenke (Autosomal-rezessive Vererbung)[24] |
| Dermatosparaxis Typ | Dermatosparaxis – EDS Typ VIIC | COL1 pNPI Mutation | Haut sehr schlaff, deutliche Überbeweglichkeit der Gelenke, Beteiligung der inneren Organe (Autosomal-rezessive Vererbung)[24] |
| Andere, exotische Formen | X-linked EDS – EDS Typ V (X-gebunden-rezessive Vererbung)[24] | ||
| Periodontitis Typ – EDS Typ VIII (Autosomal-dominanter Erbgang)[24] | |||
| Familiäres Hypermobilitätssyndrom – EDS Typ XI | |||
| Fibronectin-deficient Typ – EDS Typ X (Autosomal-rezessive Vererbung)[24] | |||
| Progeroid EDS | B3GALT6 Mutation[41] | ||
| Nichtspezifische Formen |
Krankheitshäufigkeit
Eine Krankheit oder Erkrankung wird in Europa als selten definiert, wenn weniger als einer von zweitausend Menschen von ihr betroffen ist. EDS als Gruppe ist mit einer Rate von 1:5.000 eine der seltenen Krankheiten.[42]
Beim klassischen Typ von EDS wird das Vorkommen mit ca. 1:25.000 geschätzt.[43] Der hypermobile Typ von EDS ist mit einer geschätzten Häufigkeit von 1:10.000 die am häufigsten auftretende Art von EDS.[44] Beim vaskulären Typ wird mit einem Auftreten von 1:50.000 gerechnet.[45] Die anderen Typen von EDS sind noch seltener und treten nur vereinzelt auf (weniger als hundert bekannte Fälle pro Typ weltweit).
Das klassische (cEDS), das hypermobile (hEDS), das vaskuläre (vEDS), das arthrochalasie (aEDS) und das periodontale (pEDS) Ehlers-Danlos-Syndrom sind autosomal-dominant vererbbar, was bedeutet, dass ein Kind eine 50-prozentige Wahrscheinlichkeit auf Vererbung hat, wenn ein Elternteil diese Krankheit hat. Die anderen Ehlers-Danlos-Syndrome (clEDS, cvEDS, dEDS, kEDS, BCS, spEDS, mcEDS) sind autosomal-rezessiv vererbbar. Deren Kinder können nur die Krankheit erben, wenn beide Eltern diesen Gendefekt aufweisen. Das myopathische EDS (mEDS) kann sowohl autosomal-dominant als auch autosomal-rezessiv vererbt werden. Jedes Ehlers-Danlos-Syndrom hat eine eigene Fehlbildung, das heißt, dass Betroffene nur das EDS weitervererben können, an dem sie selbst erkrankt sind. Ein Betroffener mit vaskulärem EDS kann seinem Kind so z. B. kein klassisches EDS vererben. Jedes der Ehlers-Danlos-Syndrome kann auch de novo, also durch eine Neumutation, hervorgerufen werden.[46]
Es wird darauf hingewiesen, dass alle Häufigkeitszahlen reine Hypothesen sind. Sie stützen sich nur auf registrierte Fälle. Zurzeit sind in Deutschland etwa 5.000 Menschen mit EDS bekannt,[47] es wird jedoch mit einer hohen Dunkelziffer gerechnet.
Diagnostik und Symptomatik

Die EDS-Diagnostik beginnt zunächst mit einer eingehenden ärztlichen Untersuchung. Hierbei sollte zunächst mit Hilfe des Beighton Scores die Gelenküberbeweglichkeit getestet werden. Die Haut wird in Hinblick auf Gefühl (wie teigig oder samtig) und Überdehnbarkeit untersucht. Auch eventuelle Narben sollten begutachtet werden. Außerdem sollten Patienten umfangreich zur Symptomatik befragt und die Krankenakte beachtet werden. Die erhobenen Ergebnisse werden dann mit den klinischen Kriterien der Ehlers-Danlos-Syndrome abgeglichen. Eine finale Diagnose wird durch eine Genuntersuchung gestellt.[48] Die Diagnose hypermobiles Ehlers-Danlos-Syndrom (hEDS) wird klinisch gestellt. Die Diagnosekriterien unterteilen in drei Kriterienbereiche, die alle erfüllt werden müssen. Kriterium 1 ist ein positiver Befund hinsichtlich des Beighton Scores (mindestens 5/9 bei Erwachsenen). Kriterium 2 ist unterteilt in die Bereiche A, B und C. Um Kriterium 2 zu erfüllen, müssen zwei der drei Bereiche zutreffen. A umfasst das Vorliegen von mind. 5/12 systematischen Manifestationen, B umfasst ein familiäres Auftreten von hEDS und C umfasst Schmerzen und Instabilitäten. Kriterium 3 sieht einen Ausschluss anderer Erkrankungen vor.[12]
Differentialdiagnostisch sollten u. a. folgende Erkrankungen abgeklärt werden:
- Marfan-Syndrom, Loeys-Dietz-Syndrom Q87.4
- Osteogenesis imperfecta Q87.0
- Stickler-Syndrom Q87.8
- Achondroplasie (Chondrodysplasie) Q77.4
- Rheumatoide Arthritis M06
- Fibromyalgie M79.7
- Multiple Sklerose G35
- Hypermobilitätssyndrom M35.7
- Wachstumsschmerzen R29.8
- Akrogerie E34.8
Die Diagnostizierung von seltenen Krankheiten im Allgemeinen und EDS im Besonderen wirft in der Praxis Probleme auf. Nach einer Studie im Jahr 2005 erhielten z. B. ca. 25 % der Betroffenen ihre EDS-Diagnose erst 28 Jahre nach dem Auftreten der ersten Symptome.[49]
Für eine EDS-Diagnose ist die Erstformulierung eines klinischen Verdachtes entscheidend. Ausschlaggebend ist die Präsenz von Major- und Minor-Kriterien, die eine hohe Sensitivität für die Krankheit darstellen. Eine positive Familienhistorie kann den Verdacht auf ein EDS erhärten. In der Praxis gestaltet sich die Diagnosestellung allerdings oft anders. Meist sind es verschiedene Symptome, die in Kombinationen, Schwere und Häufigkeiten des Auftretens ungewöhnlich sind und somit die Suche nach einer erklärenden Ursache initiieren. Dabei stoßen Erkrankte häufig auf Hürden im Gesundheitssystem. Die Unkenntnis über EDS von weiten Teilen der Ärzteschaft lässt die Betroffenen oft eine jahrelange Odyssee bis zur Diagnosestellung erleben.[50]
Symptomatiken nach Bereichen (unvollständig):[51][52]
| Kardiologie | Aortenaneurysma; Gefäßerweiterungen; vergrößertes Herzkranzgefäß; Mitralklappenprolaps; Chronische Cerebro-Spinale Venöse Insuffizienz;[53] Posturales orthostatisches Tachykardiesyndrom POTS |
| Rheumatologie/Orthopädie | Hypermobilität (Beighton Score ≥ 5); Senkfuß, Knickfuß, Spreizfuß; spontane Sub-/ Luxationen; Chronische Gelenk- und Muskelschmerzen; Skoliose/Kyphose; Beinlängenunterschied; Hüftdysplasie; Hernie; Kielbrust (Pectus carinatum), frühzeitige Arthrose; Degeneration der Bandscheiben; Zysten an Gelenken, Arachnodaktylie, Klinodaktylie des kleinen Fingers.[54] |
| Dermatologie/Sportmedizin | weiche, samtige Haut; dünne, durchscheinende Haut; leichte Verletzbarkeit; breite Narben; kleine Griessknoten; verzögerte Wundheilung; Hämatomneigung; häufige Verletzungen; schnelle Ermüdung; verzögerte motorische Entwicklung; Muskelhypotonie[55] |
| Zahnmedizin | hoher Gaumen; Zahnfehlstellungen; Zahnfleischrückgang; Kiefergelenkschmerzen, kraniomandibuläre Dysfunktion[56]; hypermobile Zunge; Blutungen im Mund-/ Rachenraum |
| Gastroenterologie/Innere Medizin | Eingeweidebrüche (Hernie); Gastroparese[57]; Übelkeit;[57] Abdominalschmerz;[57] Verdauungsstörungen durch Kontraktionsstörungen; Sodbrennen, Reizdarmsyndrom, Dickdarmerweiterungen, Hämorrhoiden, Gebärmuttersenkung, Mastzellaktivitätsstörung;[53] chronische Erschöpfung[58] |
| Neurologie | Chiari-Malformation;[59] Tethered cord;[60] Dysautonomie;[61] Neuralgie; Migräne; zervikale Spondylolisthesis; HWS-Erkrankung; Kopfschmerzen; generelle Koordinationsstörungen; schlechtes Balancegefühl; spinale Stenose; verminderte Wirksamkeit von Lokalanästhetikum; Schwäche; Müdigkeit; Schlafstörung |
| Psychiatrie | Depression; Angstzustand; Gesellschaftsrückzug durch Unverstandensein und Schmerzen; |
| Augenheilkunde | Lichtempfindlichkeit; Schielen; Kurzsichtigkeit; trockene Augen; Netzhautablösung; Linsenverschiebung; Keratokonus;[62] Makuladegeneration[63]; Grüner Star; Grauer Star |
Schmerzen
Schmerz als Diagnosekriterium bedarf wegen der Relevanz einer ausführlicheren Betrachtung. Starke Schmerzen, die sowohl chronisch als auch akut sein können, sind bei allen Typen des Ehlers-Danlos-Syndroms verbreitet.[64] Damit kann der Schmerz erheblichen Einfluss auf Betroffene nehmen und stark beeinträchtigend sein.[65] Er kann in Verbindung mit Hypermobilität, der Frequenz von (Sub-)Luxationen, Verletzungen des Bindegewebes und früheren Operationen stehen. Der Schmerz kann den Stütz- und Bewegungsapparat betreffen, in mehreren Körperregionen ("widespread pain") lokalisiert sein und als Myalgie, Arthralgie und/oder Neuralgie auftreten. Da die Ehlers-Danlos-Syndrome Erkrankungen des Bindegewebes sind, das sich im ganzen Körper befindet, können zahlreiche Vorgänge zu Schmerzen führen. Beim hypermobilen Ehlers-Danlos-Syndrom (hEDS) wird u. a. der Verlust von Propriozeption als ein wichtiger Faktor in der Entstehung von chronischem Schmerz gesehen. Die Bewegungen der Gelenke über das Normalmaß können zu ständig wiederkehrenden Gewebeverletzungen führen.
Meist ist die Schmerzbehandlung der Patienten nicht ausreichend. Das liegt auch am komplexen Entstehungsbild der Schmerzen, denn es geht häufig nicht um einen Schmerz, der seine Funktion verloren hat, sondern um tatsächliche, ständig wiederkehrende Verletzungen verschiedenster Strukturen.[66]
Therapie
Für die Ehlers-Danlos-Syndrome gibt es keine Heilung. Allgemein lässt sich sagen, dass eine auf den Patienten angepasste multimodale Therapie dringend zu empfehlen ist. Diese sollte aus Physiotherapie, Schmerztherapie und psychologischer Unterstützung bestehen.[66] Patienten mit dem vaskulären Typ gelten als am meisten gefährdet und sollten unter ständiger ärztlicher Überwachung stehen. Die medizinische Intervention für alle EDS-Typen ist auf eine symptomatische Therapie begrenzt, die sich in einer Reihe von Empfehlungen auflisten lässt.
Eine Überwachung des kardiovaskulären Systems, Physiotherapie, berufliche Rücksichtnahme (sofern das Nachgehen einer beruflichen Tätigkeit möglich ist), orthopädische Hilfsmittel, wie Orthesen, Bandagen, Gehstöcke, Rollatoren oder Rollstühle, können hilfreich sein. Aktivitäten mit Überstreckung bzw. Blockierung der Gelenke sollten vermieden werden. Notwendige chirurgische Eingriffe sollten mit Bedacht durchgeführt werden. Für die ggf. notwendige Anästhesietechnik gibt es verschiedene mögliche Erschwernisse, wie das Nichtansprechen auf Lokalanästhetika, schwierige Atemwegsverhältnisse, Neigung zu Gefäßeinrissen bei Anlage zentralvenöser Katheter und massive Blutungsereignisse insbesondere bei Bagatelloperationen bei EDS-Patienten mit fragilen Gefäßen.[67][68] Operative Eingriffe sollten nur in Zentren mit ausreichender Expertise in der Behandlung des EDS durchgeführt werden. Bänderraffungen o. Ä. zur Gelenkstabilisierung führen häufig nicht zum gewünschten Erfolg. In der Physiotherapie sollten Haltungstraining und Stabilitätsübungen mit dem Aufbau der dafür verantwortlichen kleinen Muskeln im Vordergrund stehen. Mit Bandagen können die empfindlichsten Stellen vor Verletzungen geschützt werden. Manche Patienten reagieren auf die Gabe von Vitamin C mit verminderter Schwellungsneigung und verbesserter Wundheilung. Versuche mit biotechnischem Hautersatz bei nicht heilenden Wunden waren bei einzelnen Patienten erfolgreich.[69]
Kinder sollten mit Informationen über EDS versorgt werden, sodass sie verstehen können, warum Kontaktsportarten oder andere belastende Freizeitbeschäftigungen vermieden werden sollten. Auch ist es wichtig, die Haltungskontrolle frühzeitig zu fördern, um Schäden durch Fehlhaltungen vorzubeugen. Familienmitglieder, Lehrer und Freunde sollten ebenfalls aktiv informiert werden, damit sie das Kind akzeptieren und ggf. fördern können.
Emotionale Unterstützung, Verhaltenstherapie und psychologische Unterstützung helfen den Betroffenen aller Subtypen, die Beeinträchtigung zu akzeptieren oder besser damit zurechtzukommen. Patientenorganisationen können dabei behilflich sein (siehe Weblinks).[70][71]
Das Ehlers-Danlos-Syndrom soll ab dem 1. Juli 2021 dem Heilmittelkatalog mit langfristigem Verordnungsbedarf hinzugefügt und vom Bundesministerium für Gesundheit umgesetzt werden. Dies erleichtert Betroffenen langfristige Verschreibungen für Therapien zu bekommen.[72]
Auflistung nach Fachgebieten:
| Kardiologie | Ultraschalluntersuchung jährlich, bei unauffälligem Befund alle 3 Jahre; Betablocker bei vergrößerter Aorta und/oder Bluthochdruck; Behandlung von POTS und kardio-vaskulären Problemen klassisch (wie bei Patienten ohne EDS) |
| Rheumatologie/Orthopädie | physiotherapeutische Begleitung; lebenslanges Training für Stabilität und Kraft (funktionales Reha-Programm), Übungen mit minimaler Gelenkbeteiligung und wenig Kraft: Balanceübungen, isometrische Übungen, Wassergymnastik, Tai Chi, Pilates, Vermeidung von Kontaktsportarten, repetitiven Bewegungsabläufen und Überdehnung beim Stretching; Gelenkschutz: Vermeidung von Übergewicht, Schuheinlagen; Bandagen, Kinesiotaping für am meisten betroffene Gelenke; Anpassung des Lebens- und Bewegungsstils; ergonomische Arbeits- und Hilfsmittel: unterstützende Matratzen und Kissen, Schreibutensilien mit Griffverstärkung, Haushaltshilfen; Vermeidung von stabilisierenden Operationen; Gelenksersatz, wenn notwendig; periodische Knochendichtemessung |
| Dermatologie/Sportmedizin | Vermeidung von Traumata/Verletzungen, ggf. tägliche Vitamin-C-Gabe; klebende Bandagen können zu Hautrissen führen; Wundverschluss ohne Spannung und in zwei Lagen mit längerer Liegezeit der Fäden, Zugpflaster gegen breite Narben |
| Zahnmedizin | Untersuchungen und Prophylaxe alle 6 Monate, vermindertes Ansprechen bei Lokalanästhesie beachten |
| Gastroenterologie/Innere Medizin | kleine Mahlzeiten; Behandlung von Magen-Darm-Problemen klassisch; besondere Überwachung in der Schwangerschaft |
| Neurologie | Schmerzbehandlung mit steroiden und nichtsteroiden Analgetika, anderen Non-Opioid-Analgetika und Opioid-Analgetika, Antikonvulsiva, Cannabinoiden,[66] Low Dose Naltrexone (LDN)[73] |
| Psychiatrie | psychologische Unterstützung; Schmerztherapie; Vermittlung von Patientenorganisationen |
| Augenheilkunde | regelmäßige Augenuntersuchung (Netzhaut) |
Sonstiges
EDS hypermobiler Typ vs. Hypermobilitätssyndrom:
Die klinischen Kriterien, die für das Hypermobilitätssyndrom und für die hypermobile Variante von EDS galten, waren unspezifisch und für beide Seiten nicht exklusiv.[74] Deshalb vertraten einige Ärzte und Wissenschaftler die Auffassung, dass das Hypermobilitätssyndrom eine milde Variante des hypermobilen Typs von EDS darstellt.[75] Im Zuge der neuen Diagnosekriterien von 2017 wurden die Kriterien für das hypermobile Ehlers-Danlos-Syndrom (hEDS) deutlich überarbeitet. Für Patienten, die diese nicht erfüllen und auch sonst keine andere Erkrankung die Symptome erklärt, wurde im Zuge der Aktualisierung der Begriff hypermobility spectrum disorders (HSD) eingeführt. HSD schließt die Lücke zwischen asymptomatischer Gelenksüberbeweglichkeit und hEDS.[76]
Methylentetrahydrofolat-Reduktase
Methylentetrahydrofolat-Reduktase (kurz MTHFR) spielt eine große Rolle in der Methylierung des Körpers, ist besonders an der Verstoffwechselung von B6, B12, Folsäure beteiligt.[77] Mutationen in diesem Gen werden mit mehreren Krankheitsbildern in Verbindung gebracht, besonders Neuralrohrdefekten,[78] Homocysteinurie,[79] und es wird spekuliert, ob es auch zu einem Phänotyp des Ehlers-Danlos-Syndroms führen kann. Genaueres hierzu ist nicht bekannt, es zeigten sich jedoch Mutationen in diesem Gen bei einigen Familien mit dem Ehlers-Danlos-Syndrom.[80][81][82]
Prognose
Der Ausblick für Menschen mit EDS hängt vom Typ ab, mit dem sie diagnostiziert worden sind. Die Symptome variieren sogar innerhalb der Subtypen und die Häufigkeit von Komplikationen ist von Patient zu Patient unterschiedlich. Manche Betroffene haben nur geringfügige Einschränkungen, während andere durch die Schwere im täglichen Leben stark eingeschränkt sind. Extreme Gelenkinstabilität, Schmerzen und Wirbelsäulendeformation können die Mobilität stark einschränken. Die meisten Betroffenen haben eine normale Lebenserwartung. Allerdings sind Patienten mit Gefäßbeteiligung einem erhöhten Risiko von schwerwiegenden Komplikationen ausgesetzt.
EDS ist ein lebenslanger Zustand mit meist progredientem Verlauf. Betroffene sehen sich mit sozialen Hindernissen wegen ihrer Krankheit konfrontiert. Einige Patienten berichten von Ängsten vor schwerwiegenden und schmerzhaften Rupturen, vor Verschlimmerung des Zustandes, vor Arbeitslosigkeit wegen ihrer physischen und emotionalen Lasten sowie vor sozialer Ausgrenzung im Allgemeinen. Eine Gentherapie oder andere Ansätze für eine Heilung sind nicht in Sicht, Studien dazu sind bisher nicht bekannt.
Ehlers-Danlos-Syndrom bei Tieren

Ein Ehlers-Danlos-Syndrom tritt auch bei Hunden und Katzen auf. Betroffene Tiere zeigen eine stark erhöhte Verletzbarkeit der Haut, wobei die Hautwunden schnell vernarben. Die Dehnbarkeit der Haut ist stark erhöht. Die Erkrankung wird rezessiv vererbt. Differentialdiagnostisch ist vor allem eine Verdünnung der Haut infolge hoher Kortisolspiegel (Cushing-Syndrom) oder Langzeittherapie mit Glucocorticoiden abzuklären. Die Diagnose wird elektronenmikroskopisch gesichert. Eine Behandlung ist nicht möglich.[83]
Ehlers-Danlos-Syndrom in Filmen und Serien
- Die scheinbar alkoholkranke Teenagerin Emma in Folge 4 der 13. Staffel (Langsamer Fall) der Serie Grey’s Anatomy hat EDS.[84]
- In Folge 18 der Staffel 7 der Serie Dr. House hat Nina ein Messie-Syndrom infolge der Fehlgeburt durch EDS.[85]
- In Folge 3 der 3. Staffel von Atlanta Medical hat der verletzte Schwerkriminelle ständige Schmerzen durch EDS.[86]
- Yvie Oddly, Gewinnerin der 11. Staffel der US-amerikanischen Reality-TV-Serie RuPaul’s Drag Race, leidet an EDS.[87]
- Die britische Miss Universe-Anwärterin Saarah Ahmed starb an EDS.[88]
Literatur
- Andreas Luttkus: Das Ehlers-Danlos-Syndrom: eine interdisziplinäre Herausforderung, Walter de Gruyter, 2011, ISBN 978-3-11-024955-2 eingeschränkte Vorschau in der Google Buchsuche
- Brat T. Tinkle, Carrie L. Atzinger: 24 Ehlers-Danlos-Syndrome in: Suzanne B. Cassidy, Judith E. Allanson: Management of Genetic Syndromes, John Wiley & Sons, ISBN 978-0-470-19141-5 eingeschränkte Vorschau in der Google Buchsuche
- Brat T. Tinkle: Issues and Management of Joint Hypermobility: A Guide for the Ehlers-Danlos Syndrome Hypermobility Type and the Hypermobility Syndrome, Left Paw Press, 2008, ISBN 978-0-9818360-1-0
Weblinks
- Mehrsprachige Informationen und weiterführende Links zum EDS im OrphaNet - dem Portal für seltene Krankheiten und Orphan Drugs
- über EDS (PDF; 270 kB) In: Der Orthopäde. (Memento vom 6. März 2006 im Internet Archive)
- Handlungsempfehlung zur Anästhesie bei Ehlers-Danlos Syndrom (deutsch)
- Hilfe, ich habe krumme Finger – oder die Lösung einer Reihe medizinischer Phänomene – EDS-Fallbeispiel (PDF; 2,2 MB)
- Ehlers-Danlos syndrome Genetic Home Reference. Federal Government. (englisch)
- Hypermobilität durch Bindegewebsschwäche
Patientenorganisationen:
Einzelnachweise
- Ehlers-Danlos syndrome. In: Genetics Home Reference. Abgerufen am 23. September 2019 (englisch).
- Seltene Erkrankungen. Bundesministerium für Gesundheit, 17. Mai 2019, abgerufen am 24. September 2019.
- L. A. Parapia, C. Jackson: Ehlers-Danlos syndrome–a historical review. In: British Journal of Haematology. Band 141, Nummer 1, April 2008, S. 32–35, ISSN 1365-2141. doi:10.1111/j.1365-2141.2008.06994.x. PMID 18324963.
- J. v. Meekeren: Een rekkelijke spanjert, Hooft-stuk 29. In Heel- en Geneeskonstige Aanmerkingen, van Job vall Meekeren, in sijn leven Heelmeester der Stadt, Admiraliteyt en ’t Gasthuys binnen Amsterdam. 1668, S. 170–172.
- A. N. Tschernogubow: Ein Fall von cutis laxae. In: Protokoly Moskowskawo Venere-ologitscheskawo i Dermatologitscheskawo Obtschestwa. 1891/1892, 1, S. 23–29 (cited by Steinmann et al. [21]).
- Edvard Ehlers: Cutis laxa. Neigung zu Haemorrhagien in der Haut, Lockerung mehrerer Artikulationen. In: Dermatologische Zeitung, Band 8, 1901, S. 173 f.
- H. Danlos: Un cas de cutis laxa avec tumeurs par contusion chronique des coudes et des genoux (xanthome juvénile pseudo-diabétique de MM Hallopeau et Macé de Lépinay). In: Bull Soc Franc Dermatol Syphiligraph (Paris). Band 19, 1908, S. 70–72.
- Peter J. De Coster (Hrsg.): Oral health in prevalent types of Ehlers–Danlos syndromes. In: Oral Pathol Med. 2005, 34, S. 298–307.
- 2017 EDS International Classification. In: The Ehlers Danlos Society. Abgerufen am 23. September 2019 (amerikanisches Englisch).
- The Types of EDS. In: The Ehlers Danlos Society. Abgerufen am 23. September 2019 (amerikanisches Englisch).
- HEDGE Study. In: The Ehlers Danlos Society. Abgerufen am 23. September 2019 (amerikanisches Englisch).
- hEDS Diagnostic Checklist. In: The Ehlers Danlos Society. Abgerufen am 23. September 2019 (amerikanisches Englisch).
- Tinke et al.: Hypermobile Ehlers–Danlos syndrome (a.k.a. Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history (pages 48–69). (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 23. September 2019 (englisch).
- Chopra et al.: Pain management in the Ehlers–Danlos syndromes (pages 212–219). (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 23. September 2019 (englisch).
- Brady et al.: Ehlers-Danlos Syndomes, rarer types. (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 23. September 2019 (englisch).
- Bowen et al.: Ehlers-Danlos Syndrom, classical type. (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 23. September 2019.
- Byers et al.: Diagnosis, natural history and management in vascular Ehlers-Danlos Syndrome. (PDF) In: Ehlers-Danlos Society. Americal Jourrnal of Medical Genetics, abgerufen am 23. September 2019.
- The Types of EDS. In: The Ehlers Danlos Society. Abgerufen am 23. September 2019 (amerikanisches Englisch).
- Ehlers-Danlos-Syndrom classical like Typ (clEDS): Labor & Diagnostik. Abgerufen am 31. Dezember 2020.
- Ehlers-Danlos-Syndrom classical like Typ (clEDS): Labor & Diagnostik. Abgerufen am 31. Dezember 2020.
- The Types of EDS. In: The Ehlers Danlos Society. Abgerufen am 31. Dezember 2020 (amerikanisches Englisch).
- Classical-like Ehlers-Danlos syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Abgerufen am 31. Dezember 2020.
- Fransiska Malfait, Richard Wenstrup, Anne De Paepe: Ehlers-Danlos Syndrome, Classic Type. In: ncbi.nlm.nih.gov. 29. Mai 2007, abgerufen am 12. Mai 2015.
- Ehlers-Danlos-Syndrom in: Peter Altmeyer: Enzyklopädie der Dermatologie, Allergologie, Umweltmedizin
- Martin Röcken: 19 Genetische Erkrankungen des Bindegewebes. Thieme, 2010, S. 200.
- Ehlers-Danlos-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- Howard P. Levy: Hypermobile Ehlers-Danlos Syndrome. In: GeneReviews®. University of Washington, Seattle 1993, PMID 20301456 (nih.gov [abgerufen am 31. Dezember 2020]).
- Hypermobile EDS Genetic Research Network. In: The Ehlers Danlos Society. Abgerufen am 31. Dezember 2020 (amerikanisches Englisch).
- The Types of EDS. In: The Ehlers Danlos Society. Abgerufen am 31. Dezember 2020 (amerikanisches Englisch).
- Ehlers-Danlos-Syndrom: Labor & Diagnostik. Gen-Panel, abgerufen am 31. Dezember 2020.
- Hypermobile Ehlers-Danlos syndrome. Genetic and Rare Diseases Information Center (GARD) – an NCATS Program, abgerufen am 31. Dezember 2020.
- Howard P Levy: Ehlers-Danlos Syndrome, Hypermobility Type. In: ncbi.nlm.nih.gov. 22. Oktober 2004, abgerufen am 28. Dezember 2014.
- S. Premalatha, K. N. Sarveswari, K. Lahiri: Reverse-Namaskar: a new sign in Ehlers-Danlos syndrome: a family pedigree study of four generations. In: Indian journal of dermatology. Band 55, Nummer 1, 2010, S. 86–91, doi:10.4103/0019-5154.60360, PMID 20418985, PMC 2856381 (freier Volltext).
- Ehlers-Danlos syndrome
- COL3A1 Symbol Report – HUGO Gene Nomenclature Committee. In: genenames.org. Abgerufen am 28. Dezember 2014.
- Andreas Luttkus: Das Ehlers-Danlos-Syndrom: Eine Interdisziplinäre Herausforderung, 2017, ISBN 3-11-047396-8.
- D. P. Germain: Ehlers-Danlos syndrome type IV. In: Orphanet Journal of Rare Diseases. Band 2, 2007, S. 32, doi:10.1186/1750-1172-2-32, PMID 17640391, PMC 1971255 (freier Volltext) (Review).
- Nevo-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation In: Orphanet Journal of Rare Diseases., Band 6, 2011, S. 46, ISSN 1750-1172. doi:10.1186/1750-1172-2-32. PMID 17640391. PMC 3135503 (freier Volltext). (Review).
- A. Kariminejad, B. Bozorgmehr, A. Khatami, M. H. Kariminejad, C. Giunta, B. Steinmann: Ehlers-Danlos Syndrome Type VI in a 17-Year-Old Iranian Boy with Severe Muscular Weakness - A Diagnostic Challenge? In: Iranian journal of pediatrics. Band 20, Nummer 3, September 2010, S. 358–362, PMID 23056730, PMC 3446046 (freier Volltext).
- M. Nakajima (Hrsg.): Mutations in B3GALT6, which Encodes a Glycosaminoglycan Linker Region Enzyme, Cause a Spectrum of Skeletal and Connective Tissue Disorders. Laboratory for Bone and Joint Diseases, Center for Integrative Medical Sciences, RIKEN, Tokyo 108-8639, The American Society of Human Genetics.
- Über seltene Krankheiten. Eurodis – Rare Diseases Europe, 9. Januar 2012.
- Fransiska Malfait: Ehlers-Danlos Syndrome, Classic Type. In: ncbi.nlm.nih.gov. 29. Mai 2007, abgerufen am 28. Dezember 2014.
- Howard P Levy: Ehlers-Danlos Syndrome, Hypermobility Type. In: ncbi.nlm.nih.gov. 22. Oktober 2004, abgerufen am 28. Dezember 2014.
- Karin Mayer: Ehlers-Danlos-Syndrom (EDS) Typ IV (Q79.6) (Memento vom 26. Februar 2014 im Internet Archive). Zentrum für Humangenetik und Laboratoriumsmedizin, Martinsried, vom 9. Januar 2012.
- The Types of EDS. In: The Ehlers Danlos Society. Abgerufen am 24. September 2019 (amerikanisches Englisch).
- Julia Sander, Werner Plötz: Krankenhaus Barmherzige Brüder München kümmert sich um Patienten mit seltenem Ehlers-Danlos-Syndrom. In: Ordenszeitschrift Misericordia. Ausgabe 11/08
- Malfait et al.: The 2017 International Classification of the Ehlers–Danlos Syndromes. (PDF) In: Ehlers-Danlos Society. American Journal of Human Genetics, 2017, abgerufen am 27. September 2019 (englisch).
- EurordisCare2: Studie über Diagnosezeiten am Beispiel von acht seltenen Krankheiten. Bericht über die Europäische Konferenz über seltene Krankheiten 2005, S. 26.
- Britta Berglund, Mattiasson Anne-Cathrine, Ingrid Randers: Dignity not fully upheld when seeking health care: experiences expressed by individuals suffering from Ehlers-Danlos syndrome. In: Disability and Rehabilitation. Band 32, Nr. 1, 2010, ISSN 0963-8288, S. 1–7, doi:10.3109/09638280903178407, PMID 19925271.
- So You Think You Might Have EDS? (Memento vom 27. August 2013 im Internet Archive) (PDF; 241 kB) Ehlers-Danlos National Foundation
- Ehlers-Danlos-Selbsthilfe e. V.: Statistischer Erhebungsbogen – Symptomkatalog 1. Auflage. September 2011.
- theilcfoundation.org (PDF; 362 kB)
- A. Castriota-Scanderbeg, B. Dallapiccola: Ehlers-Danlos Syndromes (PDF, S. 2) In: Abnormal Skeletal Phenotypes – From Simple Signs to Complex Diagnoses. Springer, S. 695.
- Robert Ploier: Differenzialdiagnosen in der Kinder- und Jugendmedizin. S. 290 eingeschränkte Vorschau in der Google-Buchsuche
- Mitakides et al.: Oral and Mandibular Manifestations in the Ehlers-Danlos Syndromes. (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 27. September 2019 (englisch).
- Fikree et al.: Gastrointestinal Involvement in the Ehlers-Danlos Syndromes. (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 27. September 2019 (englisch).
- Hakim et al.: Chronic Fatigue in Ehlers-Danlos Syndrome - Hypermobile Type. (PDF) In: Ehlers-Danlos Society. American Journal of Medical Genetics, 2017, abgerufen am 27. September 2019 (englisch).
- Chiari Malformation. (Nicht mehr online verfügbar.) In: dr-virella.com. Archiviert vom Original am 4. Dezember 2014; abgerufen am 28. Dezember 2014.
- Tethered Spinal Cord in Patients with Vascular Ehlers-Danlos Syndrome. In: ashg.org. Abgerufen am 28. Dezember 2014.
- Dysautonomia International – Underlying Causes of Dysautonomia. In: dysautonomiainternational.org. Abgerufen am 28. Dezember 2014.
- I. Robertson: Keratoconus and the Ehlers-Danlos syndrome: a new aspect of keratoconus. In: The Medical journal of Australia. Band 1, Nummer 18, Mai 1975, S. 571–573, ISSN 0025-729X. PMID 1143149.
- R. K. MISHRA, I. B. GOEL: DISCIFORM MACULAR DEGENERATION ASSOCIATED WITH EHLERS-DANLOS SYNDROME. (CUTIS HYPERELESTICA). In: Journal of the All-India Ophthalmological Society. Band 11, Dezember 1963, S. 87–95, ISSN 0044-7307. PMID 14098999.
- N. C. Voermans, H. Knoop u. a.: Pain in ehlers-danlos syndrome is common, severe, and associated with functional impairment. In: Journal of pain and symptom management. Band 40, Nummer 3, September 2010, S. 370–378, ISSN 1873-6513. doi:10.1016/j.jpainsymman.2009.12.026. PMID 20579833.
- N. C. Voermans, H. Knoop u. a.: Pain in ehlers-danlos syndrome is common, severe, and associated with functional impairment. In: Journal of pain and symptom management. Band 40, Nummer 3, September 2010, S. 370–378, ISSN 1873-6513. doi:10.1016/j.jpainsymman.2009.12.026. PMID 20579833.
- Linda Stapleford Bluestein: Pain Management in Patients With Hypermobility Disorders: Frequently Missed Causes of Chronic Pain. (PDF) In: Topics in Pain Management. 2017, abgerufen am 10. Juni 2019 (englisch).
- Ehlers–Danlos syndrome (Memento vom 15. April 2015 im Internet Archive) OrphanAnesthesia – ein Projekt der Deutschen Gesellschaft für Anästhesiologie und Intensivmedizin e. V. (PDF, deutsch und englisch)
- T. Wiesmann, M. Castori u. a.: Recommendations for anesthesia and perioperative management in patients with Ehlers-Danlos syndrome(s). In: Orphanet Journal of Rare Diseases. Band 9, 2014, S. 109, ISSN 1750-1172. doi:10.1186/s13023-014-0109-5. PMID 25053156. PMC 4223622 (freier Volltext).
- Babak Abai, Dena Thayer, Paul M. Glat: Two Cases of Traumatic Wounds in Patients With Ehlers-Danlos Syndrome Successfully Treated With a Bioengineered Skin Equivalent. In: Wounds. Band 15, Nummer 6, 2003.
- A. De Paepe, F. Malfait: Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. In: British Journal of Haematology. Band 127, Nummer 5, Dezember 2004, S. 491–500, ISSN 0007-1048. doi:10.1111/j.1365-2141.2004.05220.x. PMID 15566352. (Review).
- Elizabeth Russell: The Ehlers-Danlos Syndromes: An Overview. In: F.A.C.P. The Pain Practitioner. Band 16, Nr. 2, S. 27–39.
- Pressemitteilungen und Meldungen - Gemeinsamer Bundesausschuss. Abgerufen am 23. März 2021.
- Pradeep Chopra, Brad Tinkle, Claude Hamonet, Isabelle Brock, Anne Gompel: Pain management in the Ehlers–Danlos syndromes. In: American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Band 175, Nr. 1, 2017, ISSN 1552-4876, S. 212–219, doi:10.1002/ajmg.c.31554.
- L. Remvig, D. V. Jensen, R. C. Ward: Are diagnostic criteria for general joint hypermobility and benign joint hypermobility syndrome based on reproducible and valid tests? A review of the literature. In: The Journal of Rheumatology. Band 34, 2007, S. 798–803, ISSN 0315-162X. PMID 17295436. (Review).
- R. Keer, R. Grahame: Hypermobility Syndrom – Recognition and Management for Physiotherapists. ISBN 978-0-7506-5390-9, S. 20.
- Irman Forghani: Updates in Clinical and Genetics Aspects of Hypermobile Ehlers Danlos Syndrome. In: Balkan Medical Journal. Band 36, Nr. 1, Januar 2019, ISSN 2146-3123, S. 12–16, doi:10.4274/balkanmedj.2018.1113, PMID 30063214, PMC 6335943 (freier Volltext).
- I. Bjelland, G. S. Tell u. a.: Folate, vitamin B12, homocysteine, and the MTHFR 677C->T polymorphism in anxiety and depression: the Hordaland Homocysteine Study. In: Archives of general psychiatry. Band 60, Nummer 6, Juni 2003, S. 618–626, ISSN 0003-990X. doi:10.1001/archpsyc.60.6.618. PMID 12796225.
- L. Yan, L. Zhao u. a.: Association of the maternal MTHFR C677T polymorphism with susceptibility to neural tube defects in offsprings: evidence from 25 case-control studies. In: PloS one. Band 7, Nummer 10, 2012, S. e41689, ISSN 1932-6203. doi:10.1371/journal.pone.0041689. PMID 23056169. PMC 3463537 (freier Volltext).
- Homocystinurie durch Methylen-Tetrahydrofolat-Reduktase-Mangel. In: Orphanet (Datenbank für seltene Krankheiten).
- Z. H. Shagirova, L. N. Ushenkova u. a.: [Investigation of detoxification polymorphisms genes, methylenetetrahydrofolate-reductase (MTHFR) and P53 in the radiosensitive human cells]. In: Radiatsionnaia biologiia, radioecologiia. Band 50, Nummer 2, 2010 Mar-Apr, S. 128–133, ISSN 0869-8031. PMID 20464958.
- Talking with MTHFR Expert Dr. Ben Lynch – Show Timeline. In: gapsdietjourney.com. 11. Juni 2012, abgerufen am 28. Dezember 2014.
- mthfrheds.com. 12. Februar 2010, abgerufen am 28. Dezember 2014.
- Ch. Noli, F. Scarampella: Ehlers-Danlos-Syndrom. In: Praktische Dermatologie bei Hund und Katze. Schlütersche Verlagsanstalt, 2. Aufl. 2005, S. 331–332. ISBN 3-87706-713-1
- Falling Slowly. Abgerufen am 1. Juli 2020 (englisch).
- Dr. House: #7.18 Verschüttete Wahrheiten. myFanbase, abgerufen am 1. Juli 2020.
- Atlanta Medical – Staffel 3 Episode 3: Heilige Sünderin. 24. Juni 2020, abgerufen am 1. Juli 2020 (österreichisches Deutsch).
- Yvie Oddly wins RuPaul’s Drag Race 11 and the hearts of the EDS community. In: The Ehlers Danlos Society. 30. Mai 2019, abgerufen am 27. Juli 2020 (amerikanisches Englisch).
- FOCUS Online: Britische Miss-Universe-Anwärterin stirbt mit nur 20 Jahren an seltener Krankheit. Abgerufen am 19. April 2021.