Proteinkristall
Proteinkristalle werden in der Proteinkristallographie untersucht, sie bestehen aus gereinigtem Protein und großen Anteilen von Kristallwasser.[1] In einem Proteinkristall sind, wie bei jedem Kristall aus organischen oder anorganischen Verbindungen, identische Moleküle oder Molekularkomplexe an den Gitterpunkten des Kristallgitters exakt gleich angeordnet.[1] Die Basis des Kristalls besteht dann aus bis zu einigen tausend Atomen. Die Strukturaufklärung von Proteinen liefert nicht nur wertvolle Informationen für die Grundlagenforschung, sondern unterstützt auch zunehmend die Entwicklung von Medikamenten in der Pharmazeutischen Industrie.




Geschichte
Während man bei Kristallen von einfach gebauten chemischen Verbindungen mit der noch jungen Methode der Röntgenstrukturanalyse relativ rasch die Struktur aufklären konnte (Natriumchlorid 1913, Benzol 1928), traten große experimentelle Schwierigkeiten bei Proteinen auf, da diese aus tausenden Atomen bestehen. Die erste Schwierigkeit war die Isolierung, Reinigung und Kristallisation von Proteinen. Dies gelang James Batcheller Sumner erstmals bei dem Enzym Urease 1926 und bei den Proteinen Concanavalin A und B aus der Jackbohne. Die Anwendung der Proteinkristallisation als allgemeine Methode konnte John Howard Northrop z. B. anhand des Pepsins 1929 zeigen. Beide Forscher erhielten für diese Entwicklungen im Jahre 1946 den Nobelpreis für Chemie.
Erst Anfang der 1930er Jahre gelang es dem britischen Physiker, Kristallographen und Wissenschaftshistoriker John Desmond Bernal und seiner Mitarbeiterin Dorothy Crowfoot Hodgkin (die 1964 alleine den Nobelpreis für Chemie erhielt), von Proteinkristallen scharfe Beugungsbilder zu erhalten. Um aus diesen Beugungsbildern die dreidimensionale Proteinstruktur zu ermitteln, war jedoch ein großer rechnerischer Aufwand erforderlich, der erst mit der Entwicklung des Computers einfacher zu bewältigen war. Die erste Struktur eines Proteins (Myoglobin) wurde von John Cowdery Kendrew mit Hilfe der Röntgenkristallographie 1958 aufgeklärt (Nobelpreis für Chemie 1962, gemeinsam mit Max Perutz, der die Methode entscheidend entwickelt hatte).[2] Durch das Aufkommen von Methoden zur gentechnischen Herstellung rekombinanter Proteine in den 1980er Jahren wurde die Gewinnung von Proteinen erleichtert. Zuvor stellte die Reinigung und Charakterisierung eines einzelnen Proteins noch etwa den Umfang einer biochemischen Doktorarbeit dar. Durch die gentechnischen Methoden konnten viele Proteine in deutlich erhöhten Mengen hergestellt und gereinigt werden, was den Aufwand zur Reinigung minderte. Ein weiterer Meilenstein war die Strukturaufklärung des ersten Membranproteins (Photosynthetisches Reaktionszentrum) durch Johann Deisenhofer, Robert Huber und Hartmut Michel, die 1988 mit dem Nobelpreis für Chemie geehrt wurden. Mittlerweile sind Zehntausende von Proteinen und Proteinkomplexen – auch große Partikel wie Ribosomen (erstmals durch Ada Yonath) und Viren – kristallisiert und strukturell charakterisiert worden (siehe Weblink RCSB-PDB).
Eigenschaften
Proteinkristalle entstehen nur sehr selten im Zytoplasma. Ein bemerkenswerter Unterschied zwischen Kristallen kleiner Moleküle und makromolekularen Kristallen ist der sehr große Lösungsmittelanteil in letzterem. Das Kristallwasser der Proteinkristalle nimmt etwa 30–80 % des Kristallvolumens ein und liegt teilweise in quasi-flüssiger Form in den Hohlräumen zwischen den Proteinmolekülen vor. Nur ein Bruchteil der Proteinoberfläche ist an Kristallkontakten beteiligt, der Rest ist vollständig solvatisiert, infolgedessen sind Proteinkristalle im Vergleich zu Ionen- oder Molekülkristallen sehr weich und zerbrechlich. Proteinkristalle reagieren im Gegensatz zu Ionen- oder Molekülkristallen sehr empfindlich auf Wasserverlust. Durch den hohen Wassergehalt ist es aber auch möglich, niedermolekulare Liganden, Co-Faktoren und Substrate aus der umgebenden Mutterlauge in die Lösungsmittelkanäle in den Kristall diffundieren zu lassen.[1]
Die großen Hohlräume zwischen den Proteinmolekülen lassen sich durch die Tatsache veranschaulichen, dass z. B. ein farbloser Lysozym-Kristall durch Zugabe von Methylenblau durchgehend blau gefärbt wird, den Farbstoff aber, nach Überführung in ein farbstofffreies Medium, langsam wieder durch Diffusion freisetzt (analog zu Zeolithen). Diese Eigenart von Proteinkristallen nutzt man auch in der Aufklärung der Proteinstruktur durch Kristallstrukturanalyse mittels Röntgenbeugung, indem man Schweratomderivate (z. B. Uran, Quecksilber u. a.) durch Einlegen der Kristalle in entsprechende Schwermetallsalzlösungen herstellt. Aufgrund der chiralen Natur der natürlich vorkommenden Proteine kristallisieren sie nur in den 65 chiralen[3][4] der 230 möglichen kristallographischen Raumgruppen, die keine Spiegelebenen oder Inversionszentren aufweisen.
Herstellung
Um ausreichend Protein zu erhalten, werden heute Proteine häufig zuerst in E. coli oder Hefe überexprimiert, da die Aufreinigung der Proteine wesentlich einfacher ist und man auch sehr leicht an große Mengen an Protein gelangen kann. Zunehmend werden Proteine für die Kristallisation in Insektenzellkulturen und menschlichen HEK-Zellen exprimiert. Durch die posttranslationalen Modifikationen der höheren Eukaryoten wird die korrekte Faltung mancher Proteine erst ermöglicht, was speziell für die Produktion von Membranproteinen unerlässlich sein kann.[5]
Die Grundvoraussetzung zur Herstellung von Proteinkristallen sind ausreichende Mengen an hochreinem Protein. Die Proteinmoleküle können mittels Kombination von Fällungsmethoden, Chromatographie oder präparativer Elektrophorese von anderen Proteinen getrennt werden. Die Kristallisationsbedingungen findet man anschließend, indem man hochkonzentrierte Proteinlösungen (ca. 2–20 mg Protein/ml) mit verschiedenen Puffer-Lösungen, die zumeist sehr hohe Konzentrationen an Salzen (z. B. Ammoniumsulfat), Alkoholen (z. B. Ethanol und Methylpentandiol) oder Polyethylenglykol (PEG) enthalten, in kleinen Tropfen mit Volumina im Nano- bis Mikroliterbereich vermischt und über Tage bis Wochen und Monate bei konstanter Temperatur stehen lässt.[1]
Damit Keime entstehen, muss sich das Protein-Fällungsmittelgemisch im Nukleationsbereich befinden, d. h. im Phasendiagramm im übersättigten Bereich. Folgende Methoden kommen in Betracht, sich diesem Bereich allmählich zu nähern:
- Hanging- oder Sitting-drop Methode: Ein Tropfen der Proteinlösung mit einer niedrigen Konzentration an Fällungsmitteln befindet sich oben oder seitlich in einem Gefäß über einer Lösung, die eine hohe Konzentration des Fällungsmittels aufweist. Über die Gasphase findet allmählich eine Diffusion des Lösungsmittels (Wasser) statt, welche zu einer Übersättigung im Tropfen führt.
- Diffusion über die Phasengrenze: Proteinlösung und Fällungsmittellösung werden in einer Kapillare über eine gemeinsame Phasengrenze miteinander in Kontakt gebracht. Das Fällungsmittel mit seiner viel kleineren Teilchengröße diffundiert dabei durch die Grenzfläche in die Proteinlösung.
Beim Batch-Verfahren muss sich die Lösung bereits im Nukleationsbereich befinden. Die Probe und das Fällungsmittelgemisch werden unter einer isolierenden Ölschicht miteinander zu einem Tropfen vermischt.[6]
Für die Kristallstrukturaufklärung benötigt man Einkristalle (entsprechende Kristalle sind auf den Fotos 1 und 2 abgebildet) – viel häufiger jedoch als diese entstehen ein amorpher Niederschlag (Präzipitat) oder auch Kristalle, die nicht für eine derartige Untersuchung geeignet sind (Fotos 4 und 5). Sobald auch nur kleine Proteinkristalle wachsen, ist das ein großer Erfolg, denn anschließend können die Kristallisationsbedingungen optimiert werden. Für die Löslichkeit entscheidende Parameter sind der pH-Wert, „salting in“, „salting out“, Ionenstärke, organische Lösungsmittel (Dielektrizitätskonstante) und die Temperatur. Dabei sollten die Kristallisationsparameter bei unterschiedlichen Temperaturen getestet werden, etwa bei 20 °C und 4 °C.
Eine neue Messmethode mittels hochintensiver Röntgenlaser, beispielsweise am European XFEL, erlaubt die Verwendung kleinster Proteineinkristalle, die in einem Wasserstrahl in den Strahlengang gesprüht werden und einzelne Diffraktionsbilder ergeben.[7]
Allerdings gibt es auch einige Proteine, die sich nicht kristallisieren lassen, darunter bisher die meisten Membranproteine. Diese benötigen neben den üblichen Proteinfällungsmitteln meist noch Detergentien bzw. Tenside, z. B. das β-D-Octylglucosid, die einen wasserlöslichen Molekülteil und einen hydrophoben Anteil besitzen. Letzterer bindet die hydrophobe (fettlösliche) Transmembranregion, die meist aus α-Helices besteht. Eine größere Anzahl Tensidmoleküle kann dann mit den hydrophilen Anteilen das Membranprotein in wässriger Lösung halten, ohne dass es zur Aggregation und ungeordneten Ausfällung kommt. Somit sind die schon erwähnten Proteinreinigungsmethoden anwendbar, bevor verschiedene Kristallisationsbedingungen getestet werden können.
Gelegentlich beobachtet man bei Kristallisationsansätzen auch die Bildung von recht eigenartigen Kristallen, wie zum Beispiel eines Schmetterlingkristalls (Foto 5).
Analyse
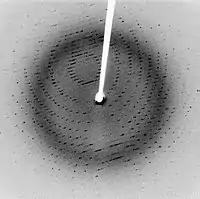
Mithilfe der Röntgenbeugung können sowohl Protein-Einkristalle (siehe Laue-Verfahren) als auch kristalline Proteinpulver (siehe Debye-Scherrer-Verfahren) untersucht werden.[1][8]
Potentielle Protein-Einkristalle werden vor der Untersuchung im Einkristalldiffraktometer meist in flüssigem Stickstoff eingefroren und dann in einem Gasstrom bei ca. −170 °C montiert, um die Schäden am Proteinkristall durch die Wechselwirkung mit der Röntgenstrahlung zu verringern. Streuende Kristalle bedeuten noch nicht, dass es sich um Proteinkristalle handelt, da aus den Puffer-Lösungen auch häufig Salze auskristallisieren. Diese kann man aber durch Untersuchung mittels eines Röntgendiffraktometers von Protein-Kristallen unterscheiden, denn beide erzeugen sehr typische, unterschiedliche Streubilder.
Für die Untersuchung mittels Röntgenbeugung sind (je nach verwendeter Methode) Einkristalle hoher Qualität und Reinheit erforderlich, deren Herstellung sehr aufwendig ist. Daher finden heutzutage vermehrt High-throughput screening bzw. automatisierte Methoden in Forschung und Industrie Anwendung, um eine große Anzahl von Proteinen zu reinigen und um Kristallisationsbedingungen zu testen. Manche Messstationen (Beamlines) an Synchrotronanlagen, die mit hochintensiver Röntgenstrahlung arbeiten, sind inzwischen mit Robotern ausgerüstet, welche das Diffraktionsverhalten von Proteinkristallen analysieren, geeignete Exemplare für die Strukturaufklärung auswählen und gegebenenfalls vollständige Diffraktionsdatensätze messen.
Neuerdings erlauben Röntgenlaser wie der European XFEL hohe Pulsraten, d. h. alle 220 Nanosekunden wird ein Röntgenblitz erzeugt, der im Idealfall das Diffraktionsbild eines im Strahlengang befindlichen Mikrokristalls erzeugt.[7] Die Methode beruht auf der kontinuierlichen Messung an vielen tausend Kristallen, deren Diffraktion schließlich einen vollständigen Datensatz von Strukturfaktoren liefert. Sobald die zugehörige Phaseninformation erhalten wird, können wie bei den eher konventionellen Methoden der Röntgenstrukturanalyse Elektronendichten berechnet werden und ein dreidimensionales Modell des Proteins mit atomaren Koordinaten erstellt werden.
Literatur
- Bernhard Rupp: Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology Garland Science, Taylor & Francis Group, New York 2010, ISBN 978-0-8153-4081-2
Weblinks
- PDBe Protein DataBank in Europe – Die Datenbank frei zugänglicher Atomkoordinaten von Biomolekülen in Europa
- PDBj Protein DataBank Japan – Die Datenbank frei zugänglicher Atomkoordinaten von Biomolekülen in Japan
- RCSB-PDB Research Collaboratory for Structural Bioinformatics – Die Datenbank frei zugänglicher Atomkoordinaten von Biomolekülen in Amerika
Einzelnachweise
- Wermuth, C. G.; Aldous, David; Raboisson, Pierre; Rognan, Didier: The practice of medicinal chemistry. 4. Auflage. London, UK, ISBN 978-0-12-417213-5.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff, D. C. Phillips: A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. In: Nature. 181, Nr. 4610, März 1958, S. 662–666. doi:10.1038/181662a0. PMID 13517261.
- Spacegroup Frequencies of PDB holdings.
- Nicolas Brener, Faiz Ben Amar, Philippe Bidaud: Designing Modular Lattice Systems with Chiral Space Groups. (PDF; 4,0 MB) 31. Oktober 2007.
- A. Pandey, K. Shin, R. E. Patterson, X. Q. Liu, J. K. Rainey: Current strategies for protein production and purification enabling membrane protein structural biology. In: Biochemistry and cell biology = Biochimie et biologie cellulaire. Band 94, Nummer 6, Dezember 2016, S. 507–527, doi:10.1139/bcb-2015-0143, PMID 27010607, PMC 5752365 (freier Volltext) (Review).
- L. Schuldt, J. Müller-Dieckmann, M. S. Weiss: Kristallisation biologischer Makromoleküle, PdN Chemie in der Schule Nr. 2/60 Jahrg. 2011, Aulis Verlag, S. 11.
- Desy News zum European XFEL bei www.desy.de, abgerufen am 19. Juli 2020.
- Macromolecular Crystallography, Teil 3, Charles W. Carter, Jr., Academic Press, 2003, ISBN 0-08-049709-8, S. 264 (eingeschränkte Vorschau in der Google-Buchsuche).