IPEX-Syndrom
Das IPEX-Syndrom ist eine sehr seltene angeborene monogenetische polyendokrine Autoimmunerkrankung. Es ist eine schwerwiegende Erkrankung, die meist schon im ersten Lebensjahr beginnt und mit einer sehr hohen Sterblichkeit verbunden ist. IPEX ist ein Akronym für Immundysregulation-Polyendokrinopathie-Enteropathie-X-chromosomal. Weitere Bezeichnungen sind beispielsweise X-chromosomal vererbte Immundysregulation, Polyendokrinopathie und Enteropathie, Immun-Dysregulation-Polyendokrinopathie-Enteropathie-Syndrom, X-chromosomal und Immundysregulation, Polyendokrinopathie und Enteropathie, X-chromosomal.[2]
| Klassifikation nach ICD-10 | |
|---|---|
| E31.0[1] | Autoimmune polyglanduläre Insuffizienz |
| ICD-10 online (WHO-Version 2019) | |
Ursache für die Erkrankung ist eine zu geringe oder vollständig fehlende körpereigene Produktion des Proteins Scurfin, auch FoxP3 (Forkhead-Box-Protein P3) genannt. Scurfin ist ein entscheidender Faktor für die Bildung von regulatorischen T-Zellen (TReg). Diese Zellen verhindern im gesunden Organismus die Entstehung von Autoimmunerkrankungen. Bei Patienten mit IPEX-Syndrom fehlen die regulatorischen T-Zellen, weshalb sich bei ihnen eine Vielzahl von Autoimmunerkrankungen gleichzeitig ausbildet. Das IPEX-Syndrom ist daher eine Multisystemerkrankung, die eine Vielzahl von Organen des Körpers betreffen kann. Abhängig von den betroffenen Organen können sehr unterschiedliche Symptome auftreten. Die individuell sehr unterschiedliche Ausprägung der Erkrankung und ihre extreme Seltenheit erschweren die Diagnose erheblich.
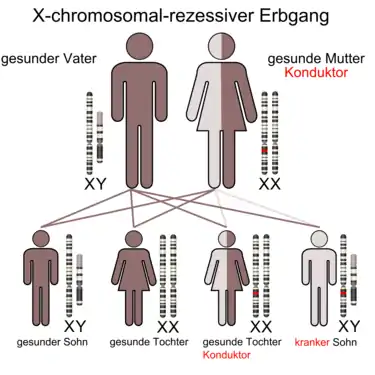
Ursache für das Defizit an FoxP3 ist in den meisten bisher bekannten Fällen eine Mutation im FOXP3-Gen, das beim Menschen auf dem X-Chromosom liegt. Bedingt durch den X-chromosomalen Erbgang erkranken am IPEX-Syndrom nur Knaben.[# 1]
Unbehandelt ist das IPEX-Syndrom innerhalb der ersten Lebensjahre tödlich. Eine Heilung ist derzeit nur durch eine hämatopoetische Stammzelltransplantation möglich.
Das IPEX-Syndrom wurde 1982 erstmals beschrieben. Bei der Scurfy-Maus, einer seit 1949 gezüchteten speziellen Mutante einer Farbmaus (Mus musculus domesticus), liegt ebenfalls eine Mutation im Foxp3-Gen vor. Die Scurfy-Maus ist mittlerweile ein wertvolles Tiermodell zur Erforschung des IPEX-Syndroms und anderer Autoimmunerkrankungen.
Epidemiologie
Das IPEX-Syndrom ist ausgesprochen selten. Weltweit sind bisher weniger als 150 Fälle erkrankter Menschen bekannt. Zuverlässige Abschätzungen über die Prävalenz wurden bisher noch nicht veröffentlicht. Allgemein wird davon ausgegangen, dass viele IPEX-Fälle nicht erkannt oder anderen Erkrankungen zugeordnet werden, die einen ähnlichen Ursprung (X-Chromosom) und ähnliche Symptome aufweisen. Dazu gehört beispielsweise das Wiskott-Aldrich-Syndrom.[3]
Genetik und Molekularbiologie
Das IPEX-Syndrom ist eine Erbkrankheit. Sie beruht auf einem durch eine Mutation hervorgerufenen Gendefekt im FOXP3-Gen, das auf dem weiblichen Geschlechtschromosom, dem X-Chromosom, liegt. Nur Frauen geben diesen Gendefekt an 50 % ihrer Nachkommen weiter. Männer erreichen, bedingt durch die hohe Sterblichkeit des IPEX-Syndroms, nicht die Geschlechtsreife. Frauen mit einem Gendefekt in FOXP3 erkranken nicht am IPEX-Syndrom. Sie sind allerdings Überträgerinnen (Konduktorinnen) der Mutation. Statistisch gesehen geben sie diese Mutation an 50 % ihrer Kinder – unabhängig vom Geschlecht – weiter. Töchter mit dieser Mutation sind wiederum nur Konduktorinnen und erkranken selbst nicht an IPEX. Ihr zweites, nicht mutiertes FOXP3-Gen auf dem zweiten X-Chromosom kompensiert den Funktionsverlust (Loss-of-Function-Mutation) des defekten Gens. Die Söhne, die diese Mutation geerbt haben, erkranken dagegen an IPEX, da männliche Individuen nur ein X-Chromosom haben.[# 2] Bei ihnen wird kein funktionsfähiges oder ausreichend funktionsfähiges Scurfin gebildet. Dieser Erbgang wird als X-chromosomal-rezessiv bezeichnet.
Das von der Mutation betroffene FOXP3-Gen liegt beim Menschen auf dem kurzen Arm des X-Chromosoms (Xp), Genlocus Xp11.23.[4] Das Genprodukt von FOXP3 (Protein) wird Scurfin oder auch FoxP3 genannt.[5] Es sind derzeit vier verschiedene Isoformen von Scurfin beim Menschen bekannt. Die Isoform 1 besteht aus 431 Aminosäuren und hat eine molare Masse von 47,2 kDa. Bei der Isoform 2 fehlen die Aminosäuren in Position 72 bis 106. Die verbliebenen 396 Aminosäuren geben dem Protein eine molare Masse von 43,4 kDa. Diese Aminosäuren fehlen auch bei der Isoform 3, allerdings sind 60 zusätzliche Aminosäuren ab Position 382 im Protein. Die molare Masse dieser aus 456 Aminosäuren bestehenden Isoform liegt bei 49,8 kDa. Bei Isoform 4 fehlen die Aminosäuren in Position 246 bis 272. Die molare Masse der verbleibenden 404 Aminosäuren liegt bei 44,4 kDa.[6] Scurfin ist ein Transkriptionsfaktor, der nach seiner Translation vor allem im Zellkern von Säugetieren vorkommt. Dort bindet Scurfin an die DNA und stimuliert die Expression von Proteinen, die in den regulatorischen T-Zellen gebraucht werden. FOXP3 ist das Meistergen (Selektorgen, engl. master gene) für regulatorische T-Zellen (TRegs).[7] Wenn naive T-Zellen, das sind nicht aktivierte T-Zellen, mit TGF-β in Kontakt kommen und gleichzeitig kein Interleukin-6 anwesend ist, so können sich diese Zellen zu TRegs differenzieren. Diese Zellen können Scurfin produzieren und durch die Ausschüttung von weiterem TGF-β, sowie von Interleukin-10 die Immunreaktion dämpfen.[8] Beides sind inhibitorische Zytokine. Fehlt funktionsfähiges Scurfin, beispielsweise durch eine Mutation im FOXP3-Gen, so wird die Immunreaktion des Organismus nicht gedämpft. Die Selbsttoleranz ist nicht mehr gegeben und es entstehen Autoimmunkrankheiten.[9] TRegs beziehungsweise FOXP3 sind essenziell für die Homöostase des Immunsystems und zur Vermeidung von Autoimmunität und überschießenden Immunreaktionen von größter Wichtigkeit.[10]
FOXP3 ist ein evolutionär hoch konserviertes Gen, das aus 12 Exons besteht.[7]
Mutationsvarianten

Die durch Mutationen hervorgerufenen Defekte im FOXP3-Gen sind sehr heterogen. Bis zum Jahr 2012 waren 63 unterschiedliche FOXP3-Mutationen bei insgesamt 136 beschrieben. Bei der Mehrzahl dieser Mutationen (27 von 63) ist der C-terminale Forkhead betroffen. Dieser Bereich ist die an die DNA bindende Proteindomäne. Die restlichen Mutationen fallen auf die Prolin-reiche Domäne (PRR) am N-Terminus (14/63), die bZIP-Domäne (5/63), die Leucin-Zipper-Forkhead-Schleife (LZ-FHK loop, 9/63), die Region oberhalb des anfänglichen ATG-Startcodons und den C-terminalen Bereich (3/63).[7]
Darüber hinaus wurden in 2 von 63 Fällen Mutationen an der Polyadenylierungsstelle (5’-AAUAAA-3’) beschrieben. Diese Mutationen führen zu einer instabilen FOXP3-mRNA. Wie bei Missense-, Frameshift-Mutationen oder Spleißdefekten, die zu einem vorzeitigen Stopcodon führen, ist in solchen Fällen ein früher und schwerwiegender Krankheitsausbruch (early-onset) sehr wahrscheinlich. Die Schwere der Erkrankung korreliert nicht in allen Fällen mit der fehlenden Expression an Scurfin. Die meisten IPEX-Patienten haben Punktmutationen, die zu einer reduzierten oder gar normalen Expression von mutiertem Scurfin führen. Allerdings ist dieses Scurfin durch Veränderung der Bindungsstellen zur DNA, der Wechselwirkung mit anderen Molekülen (beispielsweise NF-AT, AP-1 oder RORα) oder seiner Neigung zur Dimerisierung in seiner Transkriptionsregulationsaktivität beeinträchtigt.[7]
Bei der Familie, bei der 1982 erstmals das IPEX-Syndrom beschrieben wurde, konnte später keine Mutation im codierenden Bereich von FOXP3 identifiziert werden. Man geht deshalb davon aus, dass es sich hier um eine Mutation im nicht-codierenden Bereich handelt, die sich auf die Transkriptionsregulation oder das RNA-Spleißen auswirkt.[11][7]
Genotyp-Phänotyp-Korrelation
Unabhängig von der Mutationsart (dem Genotyp) ist das Erscheinungsbild (der Phänotyp) der Erkrankung bei über 90 % der Patienten durch gastrointestinale Symptome, vor allem Durchfall, gekennzeichnet. Allgemein gestaltet sich die Genotyp-Phänotyp-Korrelation beim IPEX-Syndrom schwierig, speziell wenn es um das Alter bei Krankheitsbeginn (onset) und die Einschätzung des Krankheitsverlaufs (Prognose) geht. Bei einer Gruppe von 13 Patienten liegt beispielsweise eine identische Mutation vom Typ p.Ala384Thr vor. Das heißt, an Position 384 enthält das Scurfin statt der Aminosäure Alanin die Aminosäure Threonin.[12] Bei diesen Patienten streut der Krankheitsbeginn von unmittelbar nach der Geburt (pränatal) bis in das Alter von sieben Monaten. Neben den geringen Fallzahlen erschweren therapeutische Maßnahmen und begleitende Infektionen die Korrelation zwischen Genotyp und Phänotyp.[7]
Klinisches Bild
Die meisten IPEX-Patienten werden nach einer ereignislosen Schwangerschaft von nicht miteinander verwandten Eltern geboren. Unmittelbar nach der Geburt haben sie meist ein normales Körpergewicht und eine normale Körperlänge. Pathologische Befunde liegen meist nicht vor. Die stellen sich in den ersten Lebensmonaten ein – selten in den ersten Lebenstagen oder -wochen. Wird die Krankheit nicht diagnostiziert und entsprechend behandelt, so kann sie sehr schnell zum Tod der Säuglinge führen. Die schwersten Fälle zeichnen sich durch den frühen Beginn einer Trias klinischer Manifestationen aus: hartnäckiger Durchfall, Diabetes mellitus vom Typ 1a und Ekzeme.[7]
Wesentliches Kennzeichen des IPEX-Syndroms ist die autoimmune Enteropathie: Die Patienten haben wässrigen und manchmal schleimigen (mukoiden) oder blutigen akuten Durchfall. Diese akute schwere Enteropathie ist unabhängig von der Ernährung des Säuglings (Stillen, Kuhmilch oder glutenhaltiger Nahrung). Der Durchfall führt zu einer verminderten Nährstoffausnutzung im Verdauungstrakt (Malabsorption), die Gesundheit und Entwicklung des Patienten erheblich beeinträchtigen. Oft ist deshalb eine parenterale Ernährung (künstliche Ernährung) notwendig.[7] Zusätzlich zum Durchfall kann sich das IPEX-Syndrom durch weitere gastrointestinale Störungen manifestieren. Dazu gehören: Erbrechen,[13][14][15][16] Gastritis,[17][18][19] Darmverschluss (Ileus)[20] und Kolitis (Chronisch-entzündliche Darmerkrankung).[21][16][7]
Je nach Patient kann ein Diabetes mellitus Typ 1a der Enteritis vorausgehen oder folgen. Der Diabetes ist in der Regel schwer zu kontrollieren.[22][23][18] In der überwiegenden Zahl der Fälle sind Autoantikörper nachweisbar. Es gibt seltene Fälle von Diabetes mellitus ohne Autoantikörper beim IPEX-Syndrom.[24][19] Die Zerstörung der Bauchspeicheldrüse ist durch histologische Untersuchungen, bildgebende Verfahren oder bei der Autopsie meist gut zu erkennen.[7] In der Histologie ist die massive Infiltration von Lymphozyten in dieses Organ ebenfalls gut sichtbar. Dies ist ein eindeutiges Indiz für die immunvermittelte Schädigung der Bauchspeicheldrüse.[25][24][26][7]
Diagnose
Zur Diagnosestellung werden üblicherweise die Ergebnisse der klinischen Untersuchung, die Familienanamnese und die Laborbefunde herangezogen. Molekulargenetische Tests dienen dann der endgültigen Absicherung der Diagnose[1] und sind für eine sichere Diagnosestellung unerlässlich.[18]
Die üblichen Labordaten können zu Beginn der Erkrankung weitgehend normal sein. Es gibt keine spezifischen diagnostischen Befunde für das IPEX-Syndrom. Anomale Laborwerte, die im Einklang mit dem Diabetes und der schweren Entheropatie stehen, sind der Normalfall. Autoimmunmanifestationen in anderen Zielorganen wie Hypothyreose, Zytopenien, Hepatitis oder Nephropathie können spezifisch bestimmte Laborwerte verändern. Nach Ausbruch der ersten Symptome werden bei der Mehrzahl der Patienten deutlich erhöhte Werte an Immunglobulin E und eosinophilen Granulozyten (Eosinophilie) gemessen. Die Serumspiegel an Immunglobulin A, Immunglobulin G und Immunglobulin M sind dagegen grundsätzlich normal oder – bedingt durch die Proteinverlustenteropathie – erniedrigt.[7]
Bei der Differenzialdiagnose sind in Betracht zu ziehen: das Wiskott-Aldrich- und Omenn-Syndrom, STAT-1-, CD25-, Interleukin-10-Rezeptor- oder STAT5b-Mangel, transitorischer Neugeborenen-Diabetes, Severe Combined Immunodeficiency (SCID, schwerer kombinierter Immundefekt) sowie intermediäre Formen von SCID, X-chromosomale Thrombozytopenie und Pankreashypoplasie oder Pankreasagenesie.[1]
Therapie
Kontrollierte klinische Studien mit statistisch relevanten Patientenzahlen sind aufgrund der sehr geringen Anzahl an Patienten mit IPEX-Syndrom nicht möglich. Es ist daher ausgesprochen schwierig, unterschiedliche Behandlungsmaßnahmen miteinander zu vergleichen. In der Fachliteratur werden ausschließlich Interventionen an einzelnen Patienten beschrieben (Einzelfallstudien). Aus diesen Gründen basieren alle therapeutischen Ansätze für die Behandlung des IPEX-Syndroms auf den Erfahrungen bei einzelnen Patienten. Die weitgehend unklare Genotyp-Phänotyp-Korrelation, der klinische Verlauf der Erkrankung und die Reaktion auf die Therapie können recht variabel und nicht immer befriedigend sein.[7]
Allgemeine Therapieleitlinien sind wegen der noch weitgehend unklaren Genotyp-Phänotyp-Korrelation, dem variablen klinischen Verlauf und dem unterschiedlichen Ansprechen der Patienten auf die Therapie derzeit nicht möglich. Die Therapie wird daher an den individuellen klinischen Manifestationen und ihrer Schwere ausgerichtet. Die derzeitigen Behandlungen für das IPEX-Syndrom sind die Ersatz- und unterstützende Therapie, die immunsuppressive Therapie und die hämatopoetische Stammzelltransplantation (HSZT). Mit einer Ernährungsunterstützung, beispielsweise durch parenterale Ernährung, und einer immunsuppressiven Therapie wird üblicherweise unmittelbar nach Diagnosestellung begonnen, um den akuten Manifestationen entgegenzuwirken.[7][27] Die Patienten erhalten meist eine Kombination unterschiedlicher Immunsuppressiva. Sirolimus hat sich dabei inzwischen gegenüber den Calcineurininhibitoren als vorteilhaft erwiesen. In der Langzeitnachbeobachtung konnte bei vier Patienten eine dauerhafte Remission erzielt werden.[28] Die derzeit einzige kurative Therapie ist die hämatopoetische Stammzelltransplantation. Problematisch hierbei ist jedoch, dass die Suche nach passenden Stammzellspendern in vielen Fällen erfolglos ist. Zudem sind die Patienten meist schon in einem ausgesprochen schlechten Zustand, so dass die erheblichen Nebenwirkungen der Konditionierung und Komplikationen nach der Stammzelltransplantation zu oft nicht zum gewünschten Behandlungsergebnis führen können.[29] Es hat sich jedoch gezeigt, dass bei der HSZT ein partieller Spenderchimärismus für eine vollständige Remission der Erkrankung ausreichend sein kann, wenn eine vollständige Ansiedlung (engraftment) des Treg-Kompartiments erreicht wird. Offensichtlich genügen bereits wenige funktionelle regulatorische T-Zellen zur Kontrolle der Autoimmunität beim IPEX-Syndrom.[29] An zukünftigen Behandlungsoptionen werden verschiedene Möglichkeiten der Gentherapie intensiv diskutiert und in präklinischen Studien erprobt.[29]
In klinischen Studien werden Patienten mit IPEX-Syndrom zusammen mit Patienten behandelt, die ähnliche Immundefekte, wie beispielsweise Leukozyten-Adhäsionsdefekt-Syndrome (LAD I, LAD II und LAD III), septische Granulomatose, Hyper-IgM-Syndrom Typ 1, Agammaglobulinämien (z. B. Bruton-Syndrom), Wiskott-Aldrich-Syndrom oder Chediak-Higashi-Syndrom, aufweisen.[30][31]
Prognose
Ohne eine möglichst zeitnahe Diagnose und Behandlung ist das IPEX-Syndrom meist innerhalb der ersten beiden Lebensjahre tödlich. Nach einer erfolgreichen hämatopoetischen Stammzelltransplantation ist die Lebenserwartung nahezu normal.[1]
Tiermodelle
Die Scurfy-Maus ist eine mutante Form der Hausmaus. Der Name Scurfy (engl. für „schorfig“) leitet sich von der schuppigen Haut der Tiere ab. 50 % der männlichen Nachkommen der Scurfy-Mäuse versterben im Alter von drei bis vier Wochen. Diese Tieren zeigen massive Veränderungen im Blutbild. Die Zahl an Thrombozyten und Erythrozyten ist deutlich reduziert und nimmt im Verlauf der kurzen Lebensdauer progressiv ab. Im Gegensatz dazu ist die Anzahl der Leukozyten erhöht und nimmt über die Zeit progressiv zu. Die Tiere leiden an gastrointestinalen Blutungen und Durchfall und versterben meist an einer schweren Anämie. Diese Tiere haben einen Gendefekt im foxp3-Gen, das bei Mäusen ebenfalls auf dem X-Chromosom liegt.[32] Im Gegensatz zum menschlichen Scurfin existiert das murine Scurfin in nur einer Isoform.[33]
Die Parallelen zwischen Krankheitsursache und Krankheitsverlauf bei Scurfy-Maus und Patienten mit IPEX-Syndrom machen die Scurfy-Maus zu einem ausgesprochen wertvollen Modellorganismus, nicht nur zur Erforschung des IPEX-Syndroms und der Entwicklung neuer Therapieformen, sondern ganz allgemein in dem Krankheitskomplex Autoimmunerkrankungen.[34][35][36]
Medizingeschichte

1947 wurde am Oak Ridge National Laboratory (ORNL) in Tennessee das Mammalian Genetics Laboratory gegründet. Die Aufgabe des Labors war es, die Auswirkungen von ionisierender Strahlung auf Säugetiere zu untersuchen, um daraus Rückschlüsse auf den Menschen ziehen zu können.[37] Die Leitung dieser aus dem Manhattan-Projekt abgeleiteten Forschungseinrichtung hatte William „Bill“ Russell (1910–2003). Im Rahmen dieser Studien wurden beispielsweise auch Fahrten nach New Mexico unternommen, um die Auswirkungen der Strahlung von Kernwaffenexplosionen auf Mäuse zu untersuchen. Die wissenschaftlich interessanteste Veränderung stellte sich allerdings als spontane Mutation bei einer Maus in Oak Ridge 1949 ein, die scurfy genannt wurde. Der Phänotyp, der in sich bei den Nachkommen dieser Maus einstellte, war die erste geschlechtsgebundene Erkrankung, die bei Mäusen beobachtet wurde. Zu dieser Zeit waren bei Mäusen geschlechtsgebundene Gene noch unbekannt. Die Erkrankung der betroffenen männlichen Tiere wurde durch eine letztlich für sie tödlich verlaufende rezessive Mutation hervorgerufen. Liane B. Russell (1923–2019), die Ehefrau von Bill Russell, führte an Scurfy-Mäusen eine Vielzahl bahnbrechender genetischer Versuche durch. Unter anderem erkannte sie dadurch die geschlechtsbestimmende Funktion des Y-Chromosoms.[38] Veröffentlicht wurde die Scurfy-Mutation erst 1959.[39][33]
1982 veröffentlichten Berkley R. Powell, Neil R. M. Buist und Peter Stenzel ihre Untersuchungsergebnisse an einem Patienten, in dessen Familie 17 Kinder in den ersten Lebensjahren verstarben. Dazu werteten sie außerdem die Krankenakten von acht der Verstorbenen aus. In dieser Erstbeschreibung des IPEX-Syndroms postulieren die Autoren, dass ein unbekannter genetischer Mechanismus auf dem X-Chromosom für die Überaktivität des Immunsystems verantwortlich ist.[40]
Dass Scurfy-Mäuse ähnliche Symptome aufweisen wie Patienten mit dem Wiskott-Aldrich-Syndrom beim Menschen, wurde 1990 festgestellt. Unterschiede bei einzelnen Symptomen und ein anderer Genlokus zeigten aber, dass es sich um unterschiedliche Gene handeln muss. Eine der Scurfy-Maus entsprechende menschliche Erkrankung kannten die Autoren nicht.[41] Erste immunologische Untersuchungen an Scurfy-Mäusen zeigten eine massive Infiltration von Lymphozyten und myeloischen Zellen in einer Vielzahl von Organen und machten die ausgeprägte autoimmune Pathologie deutlich.[42][43][33]
Der Genlocus Xp11.2, in dessen Bereich auch das beim Wiskott-Aldrich-Syndrom betroffene WAS-Gen liegt, wurde von einer Arbeitsgruppe um Ann O. Shigeoka 1993 als Ort mit direktem Bezug zum IPEX-Syndrom identifiziert.[44] Anfang des Jahres 2000 führte eine Arbeitsgruppe an der University of Virginia eine Kopplungsanalyse an 20 Mitgliedern einer vom IPEX-Syndrom betroffenen Sippe durch. Im Rahmen dieser Arbeiten konnte der Lokus auf eine Perizentromer-Region des X-Chromosoms im Bereich Xp11.23-q21.1 festgelegt werden. Außerdem kam die Arbeitsgruppe zu dem Ergebnis, dass das IPEX-Syndrom eine vom Wiskott-Aldrich-Syndrom unabhängige Erkrankung ist. Bei den untersuchten Patienten war in allen Fällen das WAS-Gen nicht mutiert.[13] Eine Arbeitsgruppe um Talal A. Chatila entdeckte im selben Jahr, dass das IPEX-Syndrom durch Mutationen im FOXP3-Gen, seinerzeit noch JM2-Gen bezeichnet, ausgelöst wird.[45]
Der Zusammenhang zwischen FoxP3 und der Scurfy-Maus,[46] sowie mit dem humanen neonatalen Diabetes Typ 1 wurde erstmals im Jahr 2001 festgestellt.[47] 2003 wurde die Schlüsselrolle von FoxP3 für die Bildung von regulatorischen T-Zellen[48][49] erstmals beschrieben.[47]
Literatur
- R. Bacchetta, F. Barzaghi, M. G. Roncarolo: From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. In: Annals of the New York Academy of Sciences. Band 1417, Nummer 1, April 2018, S. 5–22, doi:10.1111/nyas.13011, PMID 26918796 (Open Access).
- C. A. Chen, W. C. Chung u. a.: Quantitative analysis of tissue inflammation and responses to treatment in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, and review of literature. In: Journal of microbiology, immunology, and infection. Band 49, Nummer 5, Oktober 2016, S. 775–782, doi:10.1016/j.jmii.2015.10.015, PMID 26748735 (Review).
- Q. K. G. Tan, R. J. Louie, J. W. Sleasman: IPEX Syndrome. In: M. P. Adam, H. H. Ardinger et al.: GeneReviews. 2004, University of Washington, Seattle. PMID 20301297
Fußnoten
- Die Weitergabe des Gendefektes durch einen Vater ist durch die frühe und hohe Letalität der Erkrankung ausgeschlossen, so dass es keine homozygoten Trägerinnen der Mutation geben kann.
- Ausnahme sind Männer mit Klinefelter-Syndrom.
Einzelnachweise
- Immundysregulation-Polyendokrinopathie-Enteropathie-X-chromosomal-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- S. Daum, B. Siegmund, M. Zeitz: Immunerkrankungen des Intestinaltraks inklusive intestinaler Lymphome. In: Helmut Messmann (Hrsg.): Klinische Gastroenterologie. Georg Thieme Verlag, 2011, ISBN 978-3-131-65991-0, S. 348 (eingeschränkte Vorschau in der Google-Buchsuche).
- Mark C. Hannibal, Troy Torgerson: IPEX Syndrome. In: R. A. Pagon, M. P. Adam u. a. (Hrsg.): GeneReviews. University of Washington, Seattle, 2004 (aktualisiert 2011), PMID 20301297
- IPEX-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- R. S. Wildin, A. Freitas: IPEX and FOXP3: clinical and research perspectives. In: Journal of autoimmunity. Band 25 Suppl, 2005, S. 56–62, doi:10.1016/j.jaut.2005.04.008, PMID 16243487 (Review).
- Q9BZS1 (FOXP3_HUMAN) Universal Protein Resource (UniProt), abgerufen am 15. März 2017
- F. Barzaghi, L. Passerini, R. Bacchetta: Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. In: Frontiers in immunology. Band 3, 2012, S. 211, doi:10.3389/fimmu.2012.00211, PMID 23060872, PMC 3459184 (freier Volltext). Open Access CC-by-3.0
- Stefan Rehart: Expertise Orthopädische Rheumatologie. Georg Thieme Verlag, 2015, ISBN 978-3-131-71421-3, S. 29 (eingeschränkte Vorschau in der Google-Buchsuche).
- J. D. Fontenot, A. Y. Rudensky: A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. In: Nature immunology. Band 6, Nummer 4, April 2005, S. 331–337, doi:10.1038/ni1179, PMID 15785758 (Review).
- Fritz H. Kayser: Taschenlehrbuch Medizinische Mikrobiologie. Georg Thieme Verlag, 2014, ISBN 978-3-131-51443-1, S. 150 (eingeschränkte Vorschau in der Google-Buchsuche).
- C. L. Bennett, J. Christie u. a.: The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. In: Nature genetics. Band 27, Nummer 1, Januar 2001, S. 20–21, doi:10.1038/83713, PMID 11137993.
- NM_014009.3(FOXP3):c.1150G>A (p.Ala384Thr) AND Insulin-dependent diabetes mellitus secretory diarrhea syndrome. National Center for Biotechnology Information, abgerufen am 16. März 2017
- P. J. Ferguson, S. H. Blanton u. a.: Manifestations and linkage analysis in X-linked autoimmunity-immunodeficiency syndrome. In: American journal of medical genetics. Band 90, Nummer 5, Februar 2000, S. 390–397, PMID 10706361.
- Y. Hashimura, K. Nozu u. a.: Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. In: Pediatric nephrology. Band 24, Nummer 6, Juni 2009, S. 1181–1186, doi:10.1007/s00467-009-1119-8, PMID 19189134.
- R. Harbuz, J. Lespinasse u. a.: Identification of new FOXP3 mutations and prenatal diagnosis of IPEX syndrome. In: Prenatal diagnosis. Band 30, Nummer 11, November 2010, S. 1072–1078, doi:10.1002/pd.2613, PMID 20842625.
- K. Otsubo, H. Kanegane u. a.: Identification of FOXP3-negative regulatory T-like (CD4(+)CD25(+)CD127(low)) cells in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. In: Clinical immunology. Band 141, Nummer 1, Oktober 2011, S. 111–120, doi:10.1016/j.clim.2011.06.006, PMID 21802372.
- D. S. Nieves, R. P. Phipps u. a.: Dermatologic and immunologic findings in the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. In: Archives of dermatology. Band 140, Nummer 4, April 2004, S. 466–472, doi:10.1001/archderm.140.4.466, PMID 15096376 (Review).
- E. Gambineri, L. Perroni u. a.: Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. In: The Journal of allergy and clinical immunology. Band 122, Nummer 6, Dezember 2008, S. 1105–1112.e1, doi:10.1016/j.jaci.2008.09.027, PMID 18951619.
- M. Scaillon, S. Van Biervliet u. a.: Severe gastritis in an insulin-dependent child with an IPEX syndrome. In: Journal of Pediatric Gastroenterology and Nutrition. Band 49, Nummer 3, September 2009, S. 368–370, doi:10.1097/MPG.0b013e3181a159de, PMID 19633572.
- E. Levy-Lahad, R. S. Wildin: Neonatal diabetes mellitus, enteropathy, thrombocytopenia, and endocrinopathy: Further evidence for an X-linked lethal syndrome. In: The Journal of pediatrics. Band 138, Nummer 4, April 2001, S. 577–580, doi:10.1067/mpd.2001.111502, PMID 11295725.
- K. G. Lucas, D. Ungar u. a.: Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome. In: Bone Marrow Transplantation. Band 39, Nummer 1, Januar 2007, S. 55–56, doi:10.1038/sj.bmt.1705542, PMID 17115064.
- J. E. Peake, R. B. McCrossin u. a.: X-linked immune dysregulation, neonatal insulin dependent diabetes, and intractable diarrhoea. In: Archives of disease in childhood. Fetal and neonatal edition. Band 74, Nummer 3, Mai 1996, S. F195–F199, PMID 8777684, PMC 2528343 (freier Volltext).
- O. Baud, O. Goulet u. a.: Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. In: The New England Journal of Medicine. Band 344, Nummer 23, Juni 2001, S. 1758–1762, doi:10.1056/NEJM200106073442304, PMID 11396442.
- O. Rubio-Cabezas, J. A. Minton u. a.: Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. In: Diabetes care. Band 32, Nummer 1, Januar 2009, S. 111–116, doi:10.2337/dc08-1188, PMID 18931102, PMC 2606841 (freier Volltext).
- B. T. Costa-Carvalho, M. I. de Moraes-Pinto u. a.: A remarkable depletion of both naïve CD4+ and CD8+ with high proportion of memory T cells in an IPEX infant with a FOXP3 mutation in the forkhead domain. In: Scandinavian journal of immunology. Band 68, Nummer 1, Juli 2008, S. 85–91, doi:10.1111/j.1365-3083.2008.02055.x, PMID 18489537.
- R. S. Wildin, S. Smyk-Pearson, A. H. Filipovich: Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. In: Journal of medical genetics. Band 39, Nummer 8, August 2002, S. 537–545, PMID 12161590, PMC 1735203 (freier Volltext) (Review).
- P. L. Yong, P. Russo, K. E. Sullivan: Use of sirolimus in IPEX and IPEX-like children. In: Journal of clinical immunology. Band 28, Nummer 5, September 2008, S. 581–587, doi:10.1007/s10875-008-9196-1, PMID 18481161.
- L. Bindl, T. Torgerson u. a.: Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). In: The Journal of pediatrics. Band 147, Nummer 2, August 2005, S. 256–259, doi:10.1016/j.jpeds.2005.04.017, PMID 16126062.
- L. Passerini, F. R. Santoni de Sio u. a.: Gene/cell therapy approaches for Immune Dysregulation Polyendocrinopathy Enteropathy X-linked syndrome. In: Current gene therapy. Band 14, Nummer 6, 2014, S. 422–428, PMID 25274247, PMC 4443799 (freier Volltext) (Review).
- Klinische Studie (Phase II): Reduced Intensity Conditioning for Hemophagocytic Syndromes or Selected Primary Immune Deficiencies (BMT CTN 1204) (RICHI) bei Clinicaltrials.gov der NIH
- Klinische Studie (Phase II): Immune Disorder HSCT Protocol bei Clinicaltrials.gov der NIH
- R. S. Wildin, F. Ramsdell u. a.: X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. In: Nature genetics. Band 27, Nummer 1, Januar 2001, S. 18–20, doi:10.1038/83707, PMID 11137992.
- F. Ramsdell, S. F. Ziegler: FOXP3 and scurfy: how it all began. In: Nature Reviews Immunology. Band 14, Nummer 5, Mai 2014, S. 343–349, doi:10.1038/nri3650, PMID 24722479 (Review).
- S. T. Ju, R. Sharma u. a.: The Biology of Autoimmune Response in the Scurfy Mice that Lack the CD4+Foxp3+ Regulatory T-Cells. In: Biology. Band 1, Nummer 1, April 2012, S. 18–42, doi:10.3390/biology1010018, PMID 24832045, PMC 4011033 (freier Volltext).
- S. Aschermann, C. H. Lehmann u. a.: B cells are critical for autoimmune pathology in Scurfy mice. In: Proceedings of the National Academy of Sciences. Band 110, Nummer 47, November 2013, S. 19042–19047, doi:10.1073/pnas.1313547110, PMID 24194550, PMC 3839754 (freier Volltext).
- T. Hashimoto, H. Takahashi, S. Sakaguchi: Regulatory T cell deficiency and autoimmune skin disease: beyond scurfy mouse and IPEX. In: The Journal of allergy and clinical immunology. [elektronische Veröffentlichung vor dem Druck] September 2018, doi:10.1016/j.jaci.2018.08.028, PMID 30196121.
- W. L. Russell: X-ray-induced mutations in mice. In: Cold Spring Harbor symposia on quantitative biology. Band 16, 1951, S. 327–336, PMID 14942747.
- Bill Cabage: Scurfy's legacy. Oak Ridge National Laboratory, vom 25. Januar 2017, abgerufen am 15. März 2017
- W. L. Russell, L. B. Russell, J. S. Gower: Exceptional inheritance of a sex-linked gene in the mouse explained on the basis that the X/O sex-chromosome constitution is female. In: Proceedings of the National Academy of Sciences. Band 45, Nummer 4, April 1959, S. 554–560, PMID 16590412, PMC 222595 (freier Volltext).
- B. R. Powell, N. R. Buist, P. Stenzel: An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. In: The Journal of pediatrics. Band 100, Nummer 5, Mai 1982, S. 731–737, PMID 7040622.
- M. F. Lyon, J. Peters u. a.: The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. In: Proceedings of the National Academy of Sciences. Band 87, Nummer 7, April 1990, S. 2433–2437, PMID 2320565, PMC 53703 (freier Volltext).
- V. L. Godfrey, J. E. Wilkinson u. a.: Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. In: Proceedings of the National Academy of Sciences. Band 88, Nummer 13, Juli 1991, S. 5528–5532, PMID 2062835, PMC 51910 (freier Volltext).
- V. L. Godfrey, J. E. Wilkinson, L. B. Russell: X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. In: The American journal of pathology. Band 138, Nummer 6, Juni 1991, S. 1379–1387, PMID 2053595, PMC 1886400 (freier Volltext).
- A. O. Shigeoka, P. F. Chance u. a.: Regional localization of a novel X-linked immunodeficiency with autoimmune disease and endocrinopathies to the Wiskott-Aldrich locus Xp11.2. In: Pediat. Res. 33S, 1993, 158A
- T. A. Chatila, F. Blaeser u. a.: JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. In: The Journal of clinical investigation. Band 106, Nummer 12, Dezember 2000, S. R75–R81, doi:10.1172/JCI11679, PMID 11120765, PMC 387260 (freier Volltext).
- M. E. Brunkow, E. W. Jeffery u. a.: Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. In: Nature genetics. Band 27, Nummer 1, Januar 2001, S. 68–73, doi:10.1038/83784, PMID 11138001.
- J. M. Vaquerizas, S. K. Kummerfeld u. a.: A census of human transcription factors: function, expression and evolution. In: Nature reviews. Genetics. Band 10, Nummer 4, 2009, S. 252–263, doi:10.1038/nrg2538, PMID 19274049 (Review).
- S. Hori, T. Nomura, S. Sakaguchi: Control of regulatory T cell development by the transcription factor Foxp3. In: Science. Band 299, Nummer 5609, Februar 2003, S. 1057–1061, doi:10.1126/science.1079490, PMID 12522256.
- J. D. Fontenot, M. A. Gavin, A. Y. Rudensky: Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. In: Nature immunology. Band 4, Nummer 4, April 2003, S. 330–336, doi:10.1038/ni904, PMID 12612578.