Klinefelter-Syndrom
Das Klinefelter-Syndrom, auch Klinefelter-Reifenstein-Albright-Syndrom,[1] mit dem Karyotyp 47,XXY ist eine der häufigsten Formen angeborener Chromosomenanomalien im männlichen Geschlecht und die häufigste Ursache von Hypogonadismus. Individuen mit diesem Syndrom besitzen, abweichend vom üblichen männlichen Karyotyp (46,XY), ein zusätzliches X-Chromosom in allen (47,XXY) oder einem Teil der Körperzellen (mos 47,XXY / 46,XY).[2] Die Besonderheit wurde erstmals unter wissenschaftlichen Gesichtspunkten im Jahr 1942 von den US-Amerikanern Harry F. Klinefelter, Fuller Albright und Edward Conrad Reifenstein (1908–1975)[3][4] beschrieben.[5][6] Der zugrundeliegende Karyotyp wurde 1959 erstmals von der britischen Genetikerin Patricia A. Jacobs beschrieben.[7]
| Klassifikation nach ICD-10 | |
|---|---|
| Q98.0 | Klinefelter-Syndrom, Karyotyp 47,XXY |
| ICD-10 online (WHO-Version 2019) | |
Ursache
Die genetische Ursache wurde 1959 erkannt: Man wies eine Besonderheit in der Anzahl der Geschlechtschromosomen nach, bei der zusätzlich zum üblichen Chromosomensatz des Mannes (46,XY) mindestens ein weiteres X-Chromosom in allen (Karyotyp 47,XXY) oder einem Teil der Körperzellen vorliegt (Karyotyp 46,XY/47,XXY / = Mosaik-Form / bei 6 %). Der Grund für das Vorhandensein eines überzähligen X-Chromosoms liegt darin, dass es lange vor der Zeugung des betroffenen Jungen im Ovar der Mutter oder im Hoden des Vaters bei der Geschlechtszellenbildung während der Meiose zu einem zufälligen Nicht-Auseinanderweichen (Non-Disjunction) der Geschlechtschromosomen kam, so dass mehr als eines in die reifende Eizelle bzw. Spermienzelle gelangte.[8] Die Mosaik-Form entsteht durch Non-Disjunction der Geschlechtschromosomen während der mitotischen Teilung nach der Konzeption.[9] Das überzählige X-Chromosom stammt in ungefähr der Hälfte der Fälle von der Mutter und in der anderen Hälfte vom Vater.[10] Das mütterliche Alter ist der einzige evidenzbasierte Risikofaktor für das Klinefelter-Syndrom:[10] Ab einem mütterlichen Alter von 40 Jahren ist die Wahrscheinlichkeit, ein Kind mit dem Klinefelter-Syndrom zu bekommen, viermal so hoch wie bei Frauen unter dem 24. Lebensjahr.[11][12]
Eine andere Chromosomen-Abweichung bei Männern ist das XYY-Syndrom mit einem zusätzlichen Y-Chromosom (Karyotyp 47,XYY). Die entsprechende Erscheinung bei Frauen (Karyotyp 47,XXX) ist das Triple-X-Syndrom.
Häufigkeit
Das klassische Klinefelter-Syndrom tritt bei etwa 1–2 von 1000 männlichen Neugeborenen auf.[13][14] In Deutschland leben somit etwa 41.000–82.000 Jungen bzw. Männer mit dem Klinefelter-Syndrom. Schätzungen zufolge werden jedoch lediglich 25 % aller Personen mit dem Klinefelter-Syndrom zeitlebens diagnostiziert.[15] Hochgradigere X-Chromosomenaneuploidien (z. B. 48,XXXY, 48,XXYY oder 49,XXYYY-Polysomien) werden oft als Varianten des Klinefelter-Syndroms bezeichnet, jedoch ist ihr Auftreten weitaus seltener (1:18.000–1:100.000 der männlichen Geburten). Diese Varianten verursachen ein im Vergleich zur klassischen 47,XXY-Form schwerwiegenderes Erkrankungsbild.[16]
Merkmale
Die Chromosomenbesonderheit kann sich auf die kognitive (das Denken betreffende) und körperliche Entwicklung und Leistungsfähigkeit der Jungen und Männer mit dem Klinefelter-Syndrom auswirken, wobei nicht generell gesagt werden kann, welche Symptome sich in welcher Ausprägung bei einem Jungen bzw. Mann ausbilden. Manchmal sind auch nur wenige Symptome erkennbar.[17] Generell handelt es sich um eine Hodenunterfunktion.
Kindheit
Jungen mit dem Klinefelter-Syndrom sind bei der Geburt in der Regel ganz normal entwickelt und unterscheiden sich äußerlich nicht von anderen Kindern. Gelegentlich kann jedoch ein Hodenhochstand[17] oder eine Hypospadie (Harnröhrenfehlmündung) auf ein Klinefelter-Syndrom hinweisen. Jedoch treten ein Hodenhochstand und eine Hypospadie auch bei Kindern auf, die kein Klinefelter-Syndrom haben. Zur Entwicklung von Jungen mit dem Klinefelter-Syndrom kann keine allgemein gültige Aussage gemacht werden. Während einige Jungen eine unauffällige Entwicklung haben (ein großer Teil der Betroffenen ist ja unerkannt), ist ein kleiner Teil der Betroffenen schon in der Schule beeinträchtigt (z. B. Konzentrationsprobleme, Lernprobleme, Lese-Rechtschreibschwäche).[18] Die Jungen können ruhiger und passiver als andere Kleinkinder sein. Die Trotzphase kann weniger stark ausgeprägt sein als bei ihren Geschwistern. Unspezifische Symptome wie Antriebsarmut, Kontaktarmut, geringes Selbstvertrauen, Stimmungsschwankungen bis hin zu Wutausbrüchen und sprachliche Entwicklungsstörungen in Kombination mit schulischen Problemen können auftreten. Gegenwärtig ist nicht bekannt, warum diese Untergruppe entsprechende Probleme zeigt. Diskutiert werden beispielsweise die väterliche oder mütterliche Herkunft des überzähligen X-Chromosoms.[19] Einige Jungen zeigen auch eine etwas verzögerte motorische Entwicklung: Sitzen, Krabbeln und Laufen werden etwas verspätet erlernt; eine gewisse Ungeschicklichkeit kann bei einigen Jungen mit Klinefelter-Syndrom auffallen. Weiterhin ist das Wachstum im Kindesalter etwas schneller als bei den Altersgenossen, und die Erwachsenengröße liegt im Durchschnitt etwas über dem Durchschnitt in der Normalbevölkerung. Nach aktuellem Kenntnisstand liegt die allgemeine Intelligenz von den allermeisten Jungen bzw. Männern mit Klinefelter-Syndrom, wenn überhaupt, nur knapp unter dem Intelligenz-Niveau ihrer Geschwister. Relativ häufig ist jedoch eine Teilleistungsschwäche im sprachlichen Bereich (Verbal-IQ).[18]
Pubertät und Erwachsenenalter
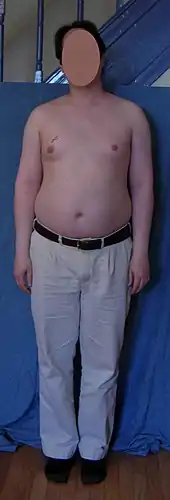
Unbehandelt kann die Pubertät bei Jungen mit Klinefelter-Syndrom verzögert eintreten und mit mehr Schwierigkeiten verbunden sein als bei anderen gleichaltrigen Jungen. Schüchternheit, Unsicherheit, eine geringe Libido und eine verminderte Wahrnehmung der eigenen Männlichkeit können zu seelischen Problemen und Isolation führen. Während bei vielen Betroffenen die Testosteronproduktion bereits während der (dann unvollständig) ablaufenden Pubertät schwindet, so tritt ein regelrechter Testosteronmangel bei den meisten Betroffenen erst im dritten Lebensjahrzehnt ein. Hinweise für einen Testosteronmangel bereits in der Pubertät können z. B. ein ausbleibender Stimmbruch sein. Anomalien der Genitalien wie ein Mikropenis, ein Scrotum bifidum oder eine Hypospadie liegen bei Individuen mit dem Klinefelter-Syndrom zwar häufiger als in der Allgemeinbevölkerung vor, jedoch liegt die diesbezügliche Prävalenz eher im niedrigen Bereich.[20] Die Hoden sind in der Regel relativ klein (ca. Größe einer Glasmurmel). Auch nach der Pubertät erhöht sich das Hodenvolumen nicht.[17] Durch den Testosteronmangel schließen sich die Wachstumsfugen der Knochen nicht rechtzeitig, sodass bei Betroffenen häufig eine überdurchschnittliche Körperhöhe mit vergleichsweise langen Beinen auftreten kann. Der fehlende oder geringe Bartwuchs kann auffallen; auch die Körperbehaarung ist in der Regel weniger ausgeprägt. Bei Personen mit dem Klinefelter-Syndrom besteht ein erhöhtes Risiko für die Entwicklung einer Gynäkomastie. Die Muskulatur ist in der Regel schwächer ausgeprägt und es lagert sich vermehrt Fettgewebe im Hüftbereich ab.
Da der größte Teil des Testosterons in den Hoden gebildet wird, besteht bei Männern mit Klinefelter-Syndrom in der Regel ein Testosteronmangel. Testosteron ist das wichtigste männliche Sexualhormon, welches eine vielfältige Rolle spielt. Es ist wichtig bei der Spermienbildung, reguliert die Libido (sexuelle Appetenz oder Sexualtrieb), ist aber auch in ganz anderen Bereichen von Bedeutung, z. B. für Fettstoffwechsel, Gefäßfunktion, Psyche, Bildung der roten Blutkörperchen, Knochenstoffwechsel und Haarwachstum (insbesondere Bart, Brust- und Genitalbehaarung). Ein Testosteronmangel kann daher verschiedenste Symptome verursachen: sexuelle Störungen (Libidoverlust, Potenzstörungen), spärlicher Bartwuchs, verringerte Muskelmasse und -kraft, verändertes Fettverteilungsmuster, Gynäkomastie und Anämie.[21] Diese Symptome können bei Personen mit dem Klinefelter-Syndrom jedoch sehr unterschiedlich ausgeprägt sein. Mit zunehmendem Alter der Betroffenen nimmt der Hypogonadismus tendenziell zu. Der Testosteronspiegel fällt im frühen Erwachsenenalter in den unteren Normalbereich. Ab einem Alter von 25 Jahren bestehen bei 65–85 % der Betroffenen klinische Zeichen eines Hypogonadismus.[17]
Eine wichtige Folge der Hodenunterentwicklung ist eine frühzeitig verringerte bzw. fehlende Spermienproduktion. Dadurch befinden sich im Ejakulat entweder zu wenige Spermien (Oligospermie) oder, wie bei den meisten Männern mit Klinefelter-Syndrom, keine Spermien (Azoospermie). Aus diesem Grund sind die allermeisten Männer mit dem Klinefelter-Syndrom unfruchtbar. Eine Ausnahme bilden Männer mit dem Mosaik-Typus (46,XY/47,XXY); bei ihnen ist die Wahrscheinlichkeit, Spermien zu finden, signifikant höher.[22]
Studien zeigen, dass Klinefelter-Patienten auch unter einem erhöhten Osteoporose-Risiko leiden. Über 40 Prozent der untersuchten Männer haben Osteoporose oder die Vorstufe Osteopenie.[23] Wie eine Studie zeigt, ist die Knochendichte in der Kindheit normal, nimmt dann aber nach der Pubertät ab.[24] Die Rolle des Testosteronmangels ist noch unklar, ein direkter Zusammenhang zwischen Testosteron und Knochendichte bei Klinefelter-Patienten wurde noch nicht beschrieben, jedoch positive Verbindungen zwischen Knochendichte, Muskelstärke (als indirekter Effekt von Testosteron) und 25(OH)-Vitamin D.[25] Neuere Studien zeigen sogar, dass die zusätzliche Therapie mit Vitamin-D-Präparaten wichtiger ist, um die Knochendichte zu erhalten, als die Testosterontherapie alleine.[26]
Begleiterkrankungen
Individuen mit dem Klinefelter-Syndrom haben im Vergleich zur Allgemeinbevölkerung ein erhöhtes Risiko für Diabetes mellitus Typ 2, Osteoporose, Gynäkomastie, Epilepsie, das Auftreten von Thrombosen und Brustkrebs.[18] Das Brustkrebsrisiko liegt jedoch unter dem normalen Risiko für Frauen. Außerdem deuten Studien auf ein erhöhtes Risiko hin, an Depressionen zu erkranken.[27]
Diagnose

Körperliche Merkmale eines Klinefelter-Syndroms können eine überdurchschnittliche Körperhöhe bis hin zu Hochwuchs, eine geringe Körperbehaarung sowie gelegentlich eine Vergrößerung der Brustdrüse sein. Es besteht in aller Regel ein kleines Hodenvolumen von 1–5 ml pro Hoden (Normwerte: 12–30 ml) und eine Unfruchtbarkeit.[18] Während der Pubertät und im Erwachsenenalter können erniedrigte Testosteronwerte mit gleichzeitig erhöhten Werten der Hypophysenhormone FSH und LH im Blut hinweisend für das Vorliegen eines Klinefelter-Syndroms sein. Auch ein Spermiogramm kann Teil der weiteren Abklärung sein. Häufig liegt eine Azoospermie, selten eine Oligospermie vor.[28] Das Klinefelter-Syndrom lässt sich jedoch nur durch eine Chromosomenanalyse zuverlässig durch den Nachweis eines zusätzlichen X-Chromosoms diagnostizieren. Als Untersuchungsmaterial genügt eine kleine Blutprobe. Um festzustellen, ob es sich um eine Mosaikform handelt, kann außerdem eine Chromosomenanalyse an Zellen der Mundschleimhaut durchgeführt werden. Viele Männer mit 47,XXY-Chromosomensatz werden im Rahmen einer medizinischen Abklärung bei unerfülltem Kinderwunsch diagnostiziert.[18] Weiterhin kann das Klinefelter-Syndrom als Zufallsbefund vorgeburtlich im Rahmen einer invasiven Pränataldiagnostik (Amniozentese, Chorionzottenbiopsie) diagnostiziert werden.
Therapie
Eine kausale Therapie ist, da das Klinefelter-Syndrom durch eine Chromosomenaberration verursacht ist, nicht möglich. Ab Beginn der Pubertät kann der bestehende Testosteronmangel durch eine entsprechende Hormon-Substitutionstherapie ausgeglichen werden.[15] Testosteronpräparate stehen in Form von Spritzen, Pflastern oder Gel zur Verfügung. Ein positiver Effekt der Testosterontherapie wurde auf neurokognitiver Ebene beim Verhalten,[29][30] dem Energieniveau,[29] dem Wohlbefinden,[29] der Lernfähigkeit[29][30] und der verbalen Fähigkeiten[31] beschrieben, während zwei andere Studien keine Auswirkungen der Testosteronbehandlung[32][33] zeigten. Ferner liegt bei Männern mit Klinefelter-Syndrom häufig ein Mangel von Vitamin D vor.[26] Daher sollte der Vitamin-D-Spiegel bei Jugendlichen und Erwachsenen regelmäßig kontrolliert werden. Insbesondere in den Wintermonaten sollte in Rücksprache mit den behandelnden Ärzten eine Vitamin-D-Substitution erfolgen, die zu einer höheren Knochendichte führt.[34][35] Sollte eine Gynäkomastie bestehen, besteht die Möglichkeit einer Mastektomie (operative Entfernung von Brustgewebe). Sofern bei Kindern eine Legasthenie, Konzentrationsschwierigkeiten oder Antriebsschwächen bestehen, besteht die Möglichkeit von Fördermaßnahmen wie beispielsweise Logopädie oder Ergotherapie. In schwerwiegenden Fällen kann ein Gespräch mit den Lehrern sinnvoll sein, um einen Nachteilsausgleich in der Schule einzufordern.
Möglichkeiten bei Unfruchtbarkeit
Die neuen Methoden der Reproduktionsmedizin, speziell die sogenannte Intrazytoplasmatische Spermieninjektion (ICSI) mit vorheriger testikulärer Spermienextraktion (TESE), haben dazu geführt, dass inzwischen wiederholt Männern mit Klinefelter-Syndrom zu leiblichen Nachkommen verholfen wurde.[36] Eine kleinere Studie ergab, dass leibliche Kinder[37] von Männern mit Klinefelter-Syndrom (Spermien durch TESE gewonnen) kein zusätzliches X-Chromosom und somit kein Klinefelter-Syndrom haben.[38] Sollten weder aus dem Sperma noch mittels der TESE funktionsfähige Samenzellen gewonnen werden können, so besteht keine Möglichkeit, leibliche Kinder zu zeugen. Da eine Testosteronbehandlung die Spermatogenese unterdrücken würde, sollte in Rücksprache mit den behandelnden Ärzten die Möglichkeit einer TESE mit dem Patienten und seinen Eltern vor Beginn der Testosterontherapie besprochen werden. Sofern bereits eine Testosterontherapie begonnen wurde, aber dennoch eine TESE gewünscht wird, so sollte die Testosterontherapie für mindestens 6 Monate pausiert werden, damit sich die Spermatogenese erholen kann.[39]
Richtlinien
September 2020 wurden von der European academy of Andrology erstmals Richtlinien für das Klinefelter-Syndrom veröffentlicht.[40]
Rolle des Androgenrezeptorgens
Das Androgenrezeptorgen (AR-Gen) liegt auf dem X-Chromosom und kodiert für den Androgenrezeptor, an dem Androgene (z. B. Testosteron) binden und wirken können. Mittels Genregulierung spielt der Androgenrezeptor eine Rolle bei der Entwicklung männlicher Geschlechtsmerkmale. Androgene und Androgenrezeptoren haben auch andere wichtige Funktionen bei Männern und Frauen, wie z. B. die Regulierung des Haarwuchses und des Sexualtriebs. In einer bestimmten Region des AR-Gens werden die DNA-Bausteine Cytosin (C), Adenin (A), Guanin (G) mehrfach wiederholt (CAGCAGCAGCAG…). Bei den meisten Menschen reicht die Anzahl der CAG-Wiederholungen („Repeats“) im AR-Gen von weniger als 10 bis etwa 36. Je kürzer dieser Repeat-Bereich ist, desto höher ist die Aktivität des Rezeptors. Studien haben ergeben, dass die Länge dieses CAG-Repeats das klinische Erscheinungsbild von Individuen mit dem Klinefelter-Syndrom beeinflusst. In einer Studie mit 77 neu diagnostizierten und unbehandelten Männern mit dem Klinefelter-Syndrom wurde eine höhere CAG-Repeatanzahl mit einer höheren Körpergröße, einer geringeren Knochendichte und einer Gynäkomastie assoziiert gefunden.[41] In einer ähnlichen Studie mit 35 Individuen mit Klinefelter-Syndrom wurde eine höhere CAG-Repeatanzahl invers mit der Penislänge korreliert vorgefunden.[42]
Störungen der geschlechtlichen Entwicklung
Die meisten Menschen werden mit 46 Chromosomen geboren. Die X- und Y-Chromosomen bestimmen das Geschlecht einer Person. Die meisten Frauen sind 46,XX und die meisten Männer sind 46,XY. Einige Individuen haben jedoch lediglich ein Geschlechtschromosom (z. B. 45,X) oder weisen mehrere Geschlechtschromosomen auf (z. B. 47,XXX; 47,XYY oder 47,XXY etc.). Darüber hinaus werden einige Männer aufgrund einer Translokation eines winzigen Abschnitts der geschlechtsdeterminierenden Region des Y-Chromosoms mit dem Chromosomensatz 46,XX geboren. Ebenso werden einige Frauen aufgrund von Mutationen im Y-Chromosom mit dem Chromosomensatz 46,XY geboren. Offensichtlich gibt es somit nicht nur 46,XX-Frauen und 46,XY-Männer, sondern es gibt eine Reihe von Chromosomenanomalien und Hormonwirkungen, die das Geschlecht mitbestimmen.[43]
Die biologischen Unterschiede zwischen Männern und Frauen resultieren aus zwei Prozessen: Geschlechtsbestimmung und -differenzierung. Der biologische Prozess der Geschlechtsbestimmung steuert, ob der männliche oder weibliche Geschlechtsdifferenzierungsweg verfolgt wird. Der Prozess der biologischen Geschlechtsdifferenzierung (Entwicklung eines bestimmten Geschlechts) umfasst viele genetisch regulierte und hierarchische Entwicklungsschritte. Wenn ein Y-Chromosom vorhanden ist, entwickeln sich um die 10. Woche der Schwangerschaft herum frühe embryonale Hoden. In Abwesenheit sowohl eines Y-Chromosoms als auch des Einflusses eines Hodenbestimmungsfaktors (TDF) entstehen Eierstöcke.[43]
Störungen der Geschlechtsentwicklung liegen definitionsgemäß dann vor, wenn chromosomales, gonadales oder phänotypisches Geschlecht nicht übereinstimmen. 2006 einigte man sich auf den Begriff „Störungen der Geschlechtsentwicklung“ (englisch Disorders of sex development, kurz DSD).[44] Man unterscheidet verschiedene DSD-Formen. Beim Klinefelter-Syndrom (47,XXY) handelt es sich um eine sogenannte DSD mit Aberration der Geschlechtschromosomen.[45] Die meisten Männer mit dem Klinefelter-Syndrom identifizieren sich mit dem männlichen Geschlecht. Jedoch gibt es auch Männer mit oder ohne Klinefelter-Syndrom, die sich eher mit dem weiblichen Geschlecht identifizieren. Es gibt in seltenen Fällen auch Zwischenformen der Geschlechtsidentität.
Die psychosexuelle Entwicklung wird in drei Kategorien eingeteilt: 1. Die Geschlechtsidentität, 2. die Geschlechterrolle (geschlechtstypisches Verhalten, welches sehr durch familiäre und gesellschaftliche Prägung beeinflusst wird) und 3. die sexuelle Orientierung. Unzufriedenheit mit dem zugewiesenen Geschlecht tritt häufiger bei Menschen mit DSD auf. Es ist schwierig zu differenzieren, was die Geschlechts-Unzufriedenheit bei Menschen mit DSD verursacht. Mögliche Ursachen umfassen eine veränderte Genregulation, eine veränderte vorgeburtliche Wirkung von Androgenen oder eine von der eigenen Wahrnehmung abweichende Geschlechtszuweisung durch familiäre oder gesellschaftliche Prägungen.[45]
Autismus und ADHS
Studien zeigten, dass bei etwa 11–12 Prozent der Personen mit Klinefelter-Syndrom die Diagnose eines Autismus gestellt wurde.[46][47] Männer mit KS wurden zuvor als schüchtern, sozial ängstlich und sozial zurückgezogen beschrieben.[48] Bruining et al.[49] untersuchten 51 Personen mit KS (6 bis 19 Jahre alt) und fanden heraus, dass 27 Prozent der Teilnehmer die Kriterien für eine Autismus-Spektrum-Störung (ASS) unter Verwendung des „Autism Diagnostic Interview-Revised“ erfüllten. Im Vergleich dazu fanden Tartaglia et al.,[50] dass 5 Prozent der Personen mit KS (6 bis 21 Jahre alt) die ASS-Kriterien unter Verwendung des „Autism Diagnostic Observation Schedule“ erfüllten. Männer mit KS haben oft verbale Schwierigkeiten, welche die soziale Kommunikation beeinträchtigen können,[51] und zum Teil mit einem erhöhten Risiko für ASS verbunden sein können.[48] Eine der führenden Klinefelter-Forscherinnen in den Niederlanden, Sophie van Rijn, beschreibt die Schwierigkeit der Betroffenen, nonverbale Signale zu erfassen und richtig zu deuten.[52]
Obwohl festgestellt wurde, dass Jungen mit dem Klinefelter-Syndrom eine erhöhte Anfälligkeit für eine Aufmerksamkeits-Defizit-Hyperaktitvitäts-Störung (ADHS) haben,[53][37] untersuchten nur wenige Studien systematisch die Auswirkungen auf die Aufmerksamkeit und Hyperaktivität bei Jungen mit KS. Tartaglia et al.[50] berichteten, dass etwa ein Drittel von 57 Männern mit KS (6 bis 21 Jahre alt) die DSM-IV-Diagnosekriterien für ADHS erfüllten. Cederlöf et al. (2013) geht von einem viermal höheren Risiko für Schizophrenie und Borderline und einem sechsmal höheren Risiko für Autismus und ADHS aus.[54]
Einordnung wissenschaftlicher Studienergebnisse
Wissenschaftliche Studien untersuchen spezifische Merkmale (z. B. Häufigkeit einer Autismus-Spektrum-Störung) bei Personen, bei denen das Klinefelter-Syndrom bereits diagnostiziert wurde. Es ist wichtig zu verstehen, dass diesbezügliche Prozentangaben nicht auf die Allgemeinheit der Individuen mit dem Klinefelter-Syndrom übertragbar sind. Diese Prozentangaben beziehen sich demnach auf die verhältnismäßig kleine Untergruppe, bei denen das Klinefelter-Syndrom bereits diagnostiziert wurde. Diese Personen hatten also spezifische Auffälligkeiten, die zu der Diagnosestellung führten. Das Klinefelter-Syndrom wird jedoch nur ca. bei 25 % der Träger im Laufe des Lebens diagnostiziert. Im Umkehrschluss bedeutet dies, dass bei 75 % der Menschen mit dem Klinefelter-Syndrom zeitlebens nicht die Diagnose gestellt wird. Da sich Studienergebnisse jedoch nur auf Personen beziehen, bei denen die Diagnose bereits gestellt wurde, sind diese häufig verzerrt und somit nicht repräsentativ. So ist beispielsweise davon auszugehen, dass eine Autismus-Spektrum-Störung oder ADHS insgesamt seltener vorliegen, als es die obigen Prozentzahlen vermuten lassen. Eine Studie wäre repräsentativ, sofern sie Individuen untersuchen würde, bei denen das Klinefelter-Syndrom bereits vorgeburtlich diagnostiziert wurde.
Wissenschaftliche Erklärungen
Körpergröße
Männer mit dem Klinefelter-Syndrom sind ca. 5–6 cm größer als Kontrollen.[55] Auf dem X- und Y-Chromosom befinden sich an den terminalen Bereichen (also an den Enden) die sogenannten pseudoautosomalen Regionen PAR1 und PAR2.[56] Gene, die sich in diesen Regionen befinden, entgehen der X-Inaktivierung und werden somit exprimiert. Bei Männern mit dem Chromosomensatz 46,XY sind demnach zwei aktive Kopien aktiv, während bei Männern mit dem Chromosomensatz 47,XXY drei Kopien aktiv sind. Innerhalb dieser pseudoautosomalen Region liegt das SHOX-Gen, welches eine wichtige Rolle beim Wachstum spielt. Männer mit dem Klinefelter-Syndrom haben somit drei aktive SHOX-Gene. Diese erhöhte Gen-Dosiswirkung ist wahrscheinlich für die hohe Statur und die langen Beine verantwortlich.[57] Die Beinlänge scheint weiterhin durch die Länge des CAG-Repeats beeinflusst zu werden. So korrelierte die Länge der CAG-Repeats positiv mit der Armlänge, der Armspanne und der Beinlänge.[55]
Siehe auch
Literatur
- J. Visootsak, J. M. Graham, Jr: Klinefelter syndrome and other sex chromosomal aneuploidies. In: Orphanet Journal of Rare Diseases. Band 1, 24. Oktober 2006, S. 42. PMID 17062147, PMC 1634840 (freier Volltext).
- E. Nieschlag: Klinefelter-Syndrom: Häufigste Form des Hypogonadismus, aber oft übersehen oder unbehandelt. In: Deutsches Ärzteblatt International. 2013; Band 110, Nr. 20, S. 347–353, doi:10.3238/arztebl.2013.0347.
- M. Bonomi, V. Rochira: Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. In: Journal of Endocrinological Investigation. 2017, Band 40, Nr. 2, S. 123–134. PMID 27644703, PMC 5269463 (freier Volltext).
Einzelnachweise
- The Klinefelter-Reifenstein-Albright syndrome. In: biomedsearch.com, abgerufen am 26. August 2017.
- Nomenklatur für den menschlichen Chromosomensatz Auf: zytogenetikforum.at.
- Dr. E. C. Reifenstein Jr., Nachruf in der New York Times, 24. Juli 1975; abgerufen am 11. August 2021.
- Anne P. Forbes, Frederic C. Bartter: Eward C. Reifenstein, Jr. In: Proceedings of the 1975 Laurentian Hormone Conference. Band 32. In: Hormone Research. 1976, S. xiii–xvii, doi:10.1016/b978-0-12-571132-6.50006-3.
- Harry F. Klinefelter Jr., Edward C. Reifenstein Jr., Fuller Albright Jr.: Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism and increased excretion of follicle-stimulating hormone. In: The Journal of Clinical Endocrinology and Metabolism, 2, Nr. 11, 1942, S. 615–624. doi:10.1210/jcem-2-11-615
- Harry F. Klinefelter Jr.: Klinefelter syndrome: historical background and development. In: Southern Medical Journal. Band 79, Nr. 9, 1986, S. 1089–1093, doi:10.1097/00007611-198609000-00012.
- P. A. Jacobs, J. A. Strong: A case of human intersexuality having a possible XXY sex-determining mechanism. In: Nature. Band 183, Nr. 4657, Januar 1959, S. 302–303. PMID 13632697.
- Klinefelter-Syndrom Genetik. Auf: klinefelter.de.
- Merel Maiburg, Sjoerd Repping, Jacques Giltay: The genetic origin of Klinefelter syndrome and its effect on spermatogenesis. In: Fertility and Sterility. Band 98, Nr. 2, August 2012, S. 253–260, doi:10.1016/j.fertnstert.2012.06.019 (elsevier.com [abgerufen am 26. September 2019]).
- George A. Kanakis, Eberhard Nieschlag: Klinefelter syndrome: more than hypogonadism. In: Metabolism. Band 86, September 2018, S. 135–144, doi:10.1016/j.metabol.2017.09.017 (elsevier.com [abgerufen am 27. November 2019]).
- Anders Bojesen, Svend Juul, Claus Højbjerg Gravholt: Prenatal and Postnatal Prevalence of Klinefelter Syndrome: A National Registry Study. In: The Journal of Clinical Endocrinology & Metabolism. Band 88, Nr. 2, Februar 2003, ISSN 0021-972X, S. 622–626, doi:10.1210/jc.2002-021491 (oup.com [abgerufen am 9. Dezember 2019]).
- F. Tuttelmann, J. Gromoll: Novel genetic aspects of Klinefelter’s syndrome. In: Molecular Human Reproduction. Band 16, Nr. 6, 1. Juni 2010, ISSN 1360-9947, S. 386–395, doi:10.1093/molehr/gaq019 (oup.com [abgerufen am 9. Dezember 2019]).
- Anders Bojesen, Svend Juul, Claus Højbjerg Gravholt: Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. In: The Journal of Clinical Endocrinology & Metabolism. Band 88, Nr. 2, 2003, S. 622–626, doi:10.1210/jc.2002-021491.
- Jeannie Visootsak, Melissa Aylstock, John M. Graham Jr.: Klinefelter syndrome and its variants: an update and review for the primary pediatrician. In: Clinical pediatrics. Band 40, Nr. 12, 2001, S. 639–651, PMID 11771918.
- Kristian A. Groth, Anne Skakkebæk, Christian Høst, Claus Højbjerg Gravholt, Anders Bojesen: Klinefelter Syndrome—A Clinical Update. In: The Journal of Clinical Endocrinology & Metabolism. Band 98, Nr. 1, Januar 2013, ISSN 0021-972X, S. 20–30, doi:10.1210/jc.2012-2382 (oup.com [abgerufen am 29. September 2019]).
- Nicole Tartaglia, Natalie Ayari, Susan Howell, Cheryl D’Epagnier, Philip Zeitler: 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome: 48, XXYY, 48, XXXY and 49, XXXXY syndromes. In: Acta Paediatrica. Band 100, Nr. 6, Juni 2011, S. 851–860, doi:10.1111/j.1651-2227.2011.02235.x, PMID 21342258, PMC 3314712 (freier Volltext).
- Christian Høst, Anne Skakkebæk, Kristian Groth, Anders Bojesen: The role of hypogonadism in Klinefelter Syndrome. In: Asian Journal of Andrology. Januar 2014, S. 185–191.
- Eberhard Nieschlag: Klinefelter Syndrome. In: Deutsches Aerzteblatt Online. 17. Mai 2013, ISSN 1866-0452, doi:10.3238/arztebl.2013.0347, PMID 23825486, PMC 3674537 (freier Volltext) – (aerzteblatt.de [abgerufen am 29. September 2019]).
- Hilgo Bruining, Sophie van Rijn, Hanna Swaab, Jacques Giltay, Wendy Kates: The Parent-of-Origin of the Extra X Chromosome May Differentially Affect Psychopathology in Klinefelter Syndrome. In: Biological Psychiatry. Band 68, Nr. 12, Dezember 2010, S. 1156–1162, doi:10.1016/j.biopsych.2010.08.034, PMID 21035791, PMC 3038433 (freier Volltext) – (elsevier.com [abgerufen am 21. Oktober 2019]).
- Yung Seng Lee, Anna Wai Fun Cheng, Syed Faisal Ahmed, Nick J. Shaw, Ieuan A. Hughes: Genital Anomalies in Klinefelter’s Syndrome. In: Hormone Research in Paediatrics. Band 68, Nr. 3, 2007, ISSN 1663-2818, S. 150–155, doi:10.1159/000106375 (karger.com [abgerufen am 9. Dezember 2019]).
- Alvaro Morales, Richard A. Bebb, Priya Manjoo, Peter Assimakopoulos, John Axler: Diagnosis and management of testosterone deficiency syndrome in men: clinical practice guideline. In: Canadian Medical Association Journal. Band 187, Nr. 18, 8. Dezember 2015, ISSN 0820-3946, S. 1369–1377, doi:10.1503/cmaj.150033, PMID 26504097, PMC 4674408 (freier Volltext) – (cmaj.ca [abgerufen am 21. Oktober 2019]).
- L. Aksglaede, A. Juul: Therapy of endocrine disease: Testicular function and fertility in men with Klinefelter syndrome: a review. (Memento vom 1. Oktober 2017 im Internet Archive) In: European Journal of Endocrinology. April 2013, Band 168, Nr. 4, S. R67–R76, doi:10.1530/EJE-12-0934.
- A. Ferlin, M. Schipilliti, C. Vinanzi, A. Garolla, A. Di Mambro u. a.: Bone mass in subjects with Klinefelter syndrome: role of testosterone levels and androgen receptor gene CAG polymorphism. In: Journal of clinical endocrinology and metabolism. Band 96, 2011, S. E739–E745. PMID 21270324.
- L. Aksglaede, C. Molgaard, N. E. Skakkebaek, A. Juul: Normal bone mineral content but unfavourable muscle/fat ratio in Klinefelter syndrome. In: Archives of Disease in Childhood. Band 93, 2008, S. 30–34, PMID 17916585.
- Christian Host u. a.: The role of hypogonadism in Klinefelter’s syndrome. In: Asian Journal of Andrology. März-April 2014, Band 16, Nr. 2, S. 185–191, PMC 3955327 (freier Volltext).
- A. Ferlin: Role of vitamin D levels and vitamin D supplementation on bone mineral density in Klinefelter syndrome. In: Osteoporosis International. August 2015, Band 26, Nr. 8, S. 2193–2202, doi: 10.1007/s00198-015-3136-8, PMID 25963234.
- z. B. H. Bruining u. a.: Psychiatric Characteristics in a self-selected sample of boys with Klinefelter Syndrome. In: Pediatrics. Band 123, 2009, S. e865.
- Anna Katharina Sophie Bade: Das Klinefelter-Syndrom: Berücksichtigung in der ärztlichen Praxis und Literatur. (PDF). 2007, abgerufen am 20. Januar 2011.
- J. Nielsen, B. Pelsen: Follow-up 20 years later of 34 Klinefelter males with karyotype 47,XXY and 16 hypogonadal males with karyotype 46,XY. In: Human Genetics. Band 77, Nr. 2, Oktober 1987, ISSN 0340-6717, S. 188–192, doi:10.1007/BF00272390 (springer.com [abgerufen am 29. September 2019]).
- Anna-Lisa Annell, Karl-Henrik Gustavson, Jan Tenstam: Symptomatology In Schoolboys With Positive Sex Chromatin (The Klinefelter Syndrome). In: Acta Psychiatrica Scandinavica. Band 46, Nr. 1, März 1970, ISSN 0001-690X, S. 71–80, doi:10.1111/j.1600-0447.1970.tb02102.x.
- A. J. Patwardhan, S. Eliez, B. Bender, M. G. Linden, A. L. Reiss: Brain morphology in Klinefelter syndrome: Extra X chromosome and testosterone supplementation. In: Neurology. Band 54, Nr. 12, 27. Juni 2000, ISSN 0028-3878, S. 2218–2223, doi:10.1212/WNL.54.12.2218.
- E. Itti, I. T. Gaw Gonzalo, A. Pawlikowska-Haddal, K. B. Boone, A. Mlikotic: The Structural Brain Correlates of Cognitive Deficits in Adults with Klinefelter’s Syndrome. In: The Journal of Clinical Endocrinology & Metabolism. Band 91, Nr. 4, April 2006, ISSN 0021-972X, S. 1423–1427, doi:10.1210/jc.2005-1596.
- Judith L. Ross, David P. Roeltgen, Gerry Stefanatos, Rebecca Benecke, Martha P.D. Zeger: Cognitive and motor development during childhood in boys with Klinefelter syndrome. In: American Journal of Medical Genetics Part A. 146A, Nr. 6, 15. März 2008, S. 708–719, doi:10.1002/ajmg.a.32232.
- A. Bojesen, N. Birkebæk, K. Kristensen, L. Heickendorff, L. Mosekilde: Bone mineral density in Klinefelter syndrome is reduced and primarily determined by muscle strength and resorptive markers, but not directly by testosterone. In: Osteoporosis International. Band 22, Nr. 5, Mai 2011, ISSN 0937-941X, S. 1441–1450, doi:10.1007/s00198-010-1354-7 (springer.com [abgerufen am 29. September 2019]).
- A. Ferlin, R. Selice, A. Di Mambro, M. Ghezzi, A. Di Nisio: Role of vitamin D levels and vitamin D supplementation on bone mineral density in Klinefelter syndrome. In: Osteoporosis International. Band 26, Nr. 8, August 2015, ISSN 0937-941X, S. 2193–2202, doi:10.1007/s00198-015-3136-8 (springer.com [abgerufen am 29. September 2019]).
- Giovanni Corona, Alessandro Pizzocaro, Fabio Lanfranco, Andrea Garolla, Fiore Pelliccione: Sperm recovery and ICSI outcomes in Klinefelter syndrome: a systematic review and meta-analysis. In: Human Reproduction Update. Band 23, Nr. 3, 1. Mai 2017, ISSN 1355-4786, S. 265–275, doi:10.1093/humupd/dmx008 (oup.com [abgerufen am 29. September 2019]).
- Martin Cederlöf, Agnes Ohlsson Gotby, Henrik Larsson, Eva Serlachius, Marcus Boman: Klinefelter syndrome and risk of psychosis, autism and ADHD. In: Journal of Psychiatric Research. Band 48, Nr. 1, Januar 2014, S. 128–130, doi:10.1016/j.jpsychires.2013.10.001 (elsevier.com [abgerufen am 4. Oktober 2019]).
- E. Greco, F. Scarselli, M. G. Minasi, V. Casciani, D. Zavaglia: Birth of 16 healthy children after ICSI in cases of nonmosaic Klinefelter syndrome. In: Human Reproduction. Band 28, Nr. 5, Mai 2013, ISSN 1460-2350, S. 1155–1160, doi:10.1093/humrep/det046.
- Akanksha Mehta, Alexander Bolyakov, Jordan Roosma, Peter N. Schlegel, Darius A. Paduch: Successful testicular sperm retrieval in adolescents with Klinefelter syndrome treated with at least 1 year of topical testosterone and aromatase inhibitor. In: Fertility and Sterility. Band 100, Nr. 4, Oktober 2013, S. 970–974, doi:10.1016/j.fertnstert.2013.06.010 (elsevier.com [abgerufen am 9. Dezember 2019]).
- Michael Zitzmann, Lise Aksglaede, Giovanni Corona, Andrea M. Isidori, Anders Juul: European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology. In: Andrology. 6. Oktober 2020, ISSN 2047-2919, S. andr.12909, doi:10.1111/andr.12909 (wiley.com [abgerufen am 24. Oktober 2020]).
- Michael Zitzmann, Marion Depenbusch, Jörg Gromoll, Eberhard Nieschlag: X-Chromosome Inactivation Patterns and Androgen Receptor Functionality Influence Phenotype and Social Characteristics as Well as Pharmacogenetics of Testosterone Therapy in Klinefelter Patients. In: The Journal of Clinical Endocrinology & Metabolism. Band 89, Nr. 12, Dezember 2004, ISSN 0021-972X, S. 6208–6217, doi:10.1210/jc.2004-1424.
- Andrew R. Zinn, Purita Ramos, Frederick F. Elder, Karen Kowal, Carole Samango-Sprouse: Androgen Receptor CAG n Repeat Length Influences Phenotype of 47,XXY (Klinefelter) Syndrome. In: The Journal of Clinical Endocrinology & Metabolism. Band 90, Nr. 9, September 2005, ISSN 0021-972X, S. 5041–5046, doi:10.1210/jc.2005-0432.
- WHO Gender and Genomics. WHO, Genomic Resource Center, abgerufen am 6. Oktober 2019.
- I A Hughes: Consensus statement on management of intersex disorders. In: Archives of Disease in Childhood. Band 91, Nr. 7, 14. Juni 2005, ISSN 0003-9888, S. 554–563, doi:10.1136/adc.2006.098319, PMID 16624884, PMC 2082839 (freier Volltext).
- Birgit Köhler: Junge oder Mädchen? Störungen der Geschlechtsentwicklung (DSD). Abgerufen am 6. Oktober 2019.
- D. V. M. Bishop, P. A. Jacobs, K. Lachlan, D. Wellesley, A. Barnicoat: Autism, language and communication in children with sex chromosome trisomies. In: Archives of Disease in Childhood. Band 96, Nr. 10, 1. Oktober 2011, ISSN 0003-9888, S. 954–959, doi:10.1136/adc.2009.179747, PMID 20656736, PMC 3182523 (freier Volltext).
- Judith L. Ross, David P. Roeltgen, Harvey Kushner, Andrew R. Zinn, Allan Reiss: Behavioral and Social Phenotypes in Boys With 47,XYY Syndrome or 47,XXY Klinefelter Syndrome. In: Pediatrics. Band 129, Nr. 4, April 2012, ISSN 0031-4005, S. 769–778, doi:10.1542/peds.2011-0719, PMID 22412026, PMC 3356148 (freier Volltext).
- Tamar Green, Shira Flash, Allan L. Reiss: Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. In: Neuropsychopharmacology. Band 44, Nr. 1, Januar 2019, ISSN 0893-133X, S. 9–21, doi:10.1038/s41386-018-0153-2, PMID 30127341, PMC 6235860 (freier Volltext) – (nature.com [abgerufen am 4. Oktober 2019]).
- H. Bruining, H. Swaab, M. Kas, H. van Engeland: Psychiatric Characteristics in a Self-Selected Sample of Boys With Klinefelter Syndrome. In: PEDIATRICS. Band 123, Nr. 5, 1. Mai 2009, ISSN 0031-4005, S. e865–e870, doi:10.1542/peds.2008-1954.
- Nicole Tartaglia, Lisa Cordeiro, Susan Howell, Rebecca Wilson, Jennifer Janusz: The Spectrum of the Behavioral Phenotype in Boys and Adolescents 47,XXY (Klinefelter Syndrome). In: Pediatric endocrinology reviews: PER. Band 8, Nr. 0 1, Dezember 2010, ISSN 1565-4753, S. 151–159, PMID 21217607, PMC 3740580 (freier Volltext).
- Jeannie Visootsak, John M. Graham: Social function in multiple X and Y chromosome disorders: XXY, XYY, XXYY, XXXY. In: Developmental Disabilities Research Reviews. Band 15, Nr. 4, 2009, S. 328–332, doi:10.1002/ddrr.76, PMID 20014367, PMC 3909519 (freier Volltext) – (wiley.com [abgerufen am 4. Oktober 2019]).
- Sophie van Rijn: One chromosome: a world of difference. 2008; abgerufen am 16. Mai 2015.
- H. Bruining, H. Swaab, M. Kas, H. van Engeland: Psychiatric Characteristics in a Self-Selected Sample of Boys With Klinefelter Syndrome. In: Pediatrics. Band 123, Nr. 5, 1. Mai 2009, ISSN 0031-4005, S. e865–e870, doi:10.1542/peds.2008-1954.
- M. Cederlöf u. a.: Klinefelter syndrome and risk of psychosis, autism and ADHD. In: Journal of Psychiatric Research. Band 48, 2013, S. 128–130.
- Simon Chang, Anne Skakkebæk, Christian Trolle, Anders Bojesen, Jens Michael Hertz: Anthropometry in Klinefelter Syndrome - Multifactorial Influences Due to CAG Length, Testosterone Treatment and Possibly Intrauterine Hypogonadism. In: The Journal of Clinical Endocrinology & Metabolism. Band 100, Nr. 3, März 2015, ISSN 0021-972X, S. E508–E517, doi:10.1210/jc.2014-2834 (oup.com [abgerufen am 9. Dezember 2019]).
- A. Helena Mangs and Brian J. Morris: The Human Pseudoautosomal Region (PAR): Origin, Function and Future. 31. März 2007, abgerufen am 9. Dezember 2019 (englisch).
- George A. Kanakis, Eberhard Nieschlag: Klinefelter syndrome: more than hypogonadism. In: Metabolism. Band 86, September 2018, S. 135–144, doi:10.1016/j.metabol.2017.09.017 (elsevier.com [abgerufen am 9. Dezember 2019]).