GABA-Rezeptor
GABA-Rezeptoren sind Transmembranproteine in Nervenzellen, die spezifisch den Neurotransmitter γ-Aminobuttersäure (GABA - Abk. v. engl. gamma-aminobutyric acid) binden. Es gibt zwei Hauptgruppen von GABA-Rezeptoren: Ionenkanal-Rezeptoren (ionotrope Rezeptoren) und Sekundärsignale auslösende (metabotrope) Rezeptoren. Zu den ionotropen Rezeptoren zählen der GABAA- und der GABAA-ρ-Rezeptor, während der GABAB-Rezeptor ein metabotroper Rezeptor ist.
GABAA-Rezeptoren
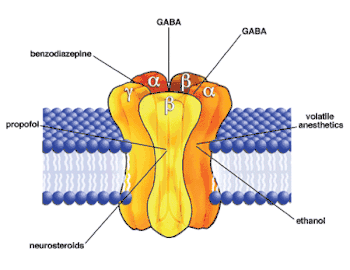
Aufbau
GABAA-Rezeptoren sind ligandenaktivierte Ionenkanäle, welche für Chlorid- und Hydrogencarbonationen durchlässig sind. Es handelt sich um Heteropentamere, das heißt, sie sind aus fünf verschiedenen Untereinheiten aufgebaut (von griechisch penta-, „fünf“, hétero, „verschieden“ und méros, „Teil“). Jede Untereinheit durchspannt die Zellmembran viermal. Es gibt acht Klassen homologer Untereinheiten.
- α (mit 6 Vertretern; α1–α6)
- β (mit 3 Vertretern; β1–β3)
- γ (mit 3 Vertretern; γ1–γ3)
- δ, ε, π, θ (mit je einem Vertreter)
- ρ (mit 3 Vertretern; ρ1–ρ3)
Die meisten GABAA-Rezeptoren im Gehirn sind aus zwei α-, zwei β- und einer γ-Untereinheiten aufgebaut. Neben der Bindestelle für γ-Aminobuttersäure besitzt der funktionsfähige GABAA-Rezeptor weitere, allosterische Bindestellen für Benzodiazepine an der γ-Untereinheit und für Barbiturate und Neurosteroide an der β-Untereinheit. Rezeptoren, die ausschließlich aus ρ-Untereinheiten zusammengesetzt sind, werden als GABAA-ρ-Rezeptoren bezeichnet.
Funktion
Der GABAA-Rezeptor ist sehr weit im Gehirn und Rückenmark verbreitet und der wichtigste inhibitorische Rezeptor im zentralen Nervensystem (ZNS). Er erhält seine Wirkung durch die Erhöhung der Permeabilität für Cl-Ionen, das ein IPSP auslöst. 30 % der Transmittermenge im ZNS entfallen auf GABA. Besondere Funktion hat er in den Basalganglien und dem Kleinhirn, wo er an der motorischen Kontrolle beteiligt ist. Die Purkinje-Zelle des Kleinhirns ist z. B. GABAerg. Im Thalamus wirkt GABA an der Einleitung und Aufrechterhaltung des Schlafs. Hier ist auch der Hauptangriffsort der pharmakologischen Beeinflussung durch Benzodiazepine und Barbiturate (s. o.). Im Rückenmark befinden sich GABA-Rezeptoren auf Motoneuronen, und sie sind an der Reflex-Verschaltung ebenso wie der Koordination von Bewegungsabläufen beteiligt (siehe: Renshaw-Zellen). Einige funktionelle Unterschiede stehen in Verbindung mit den verschiedenen α-Untereinheiten:
- α1-Untereinheiten kommen in Rezeptoren vor, welche die sedierende Wirkung von Benzodiazepinen (Diazepam) vermitteln.
- α2-Untereinheiten sind mit der anxiolytischen (angstlösenden) Funktion des Rezeptors in Verbindung zu setzen, und
- α3-Untereinheiten kommen in Rezeptoren vor, die vornehmlich eine muskelrelaxierende Wirkung besitzen.
Beeinflussung durch Alkohol
- Alkohol bindet an den GABAA-Rezeptor und führt über eine Verstärkung der Permeabilität für Chloridionen an der Nervenzellmembran zur Hyperpolarisation, wodurch die Aktionspotenzialfrequenz abnimmt. Da gleichzeitig das wichtigste exzitatorische System durch die Beeinflussung des NMDA-Rezeptors gehemmt wird, kommt es insgesamt zu einer sedierenden Wirkung.[1]
Pharmakologische Beeinflussung
- Tranquilizer wie Benzodiazepine verstärken die sedative, anxiolytische und antikonvulsive Wirkung von GABA.
- Barbiturate sind in der Lage, ohne GABA den GABAA-Rezeptor zu aktivieren, und können neben anderen Sedativa in der Anästhesie zum Einleiten der Narkose benutzt werden.
- Muscimol ist der Wirkstoff des Fliegenpilzes und ein Agonist des GABAA-Rezeptors und für dessen halluzinogene Wirkung verantwortlich.
- Bicucullin ist ein Antagonist des GABAA-Rezeptors. Dieser Wirkstoff besitzt nur experimentelle, aber keine klinische Relevanz.
- α-Thujon ist möglicherweise ein nicht-kompetitiver Antagonist am GABAA-Rezeptor.[2] Darauf beruht möglicherweise seine krampfauslösende Wirkung.
- Picrotoxin blockiert den Chloridkanal des GABAA-Rezeptors.
- Antiepileptika wie Valproat hemmen unter anderem auch den Abbau von GABA und werden verordnet, um das Auftreten von epileptischen Anfällen zu verhindern.
- Tiagabin ist ein selektiver GABA-Wiederaufnahmehemmer und soll ebenfalls die Konzentration von GABA im synaptischen Spalt erhöhen, um das Auftreten der Epilepsie zu verhindern.
- Neurosteroide sind Abbauprodukte von Androgenen und Progesteronen. Sie kommen physiologisch im Körper vor und üben eine modulierende Wirkung auf GABAA-Rezeptoren aus.
- Alphaxalon ist ein synthetisch hergestelltes Neurosteroid und wird in der Analgesie verwendet.
- Diazepam-binding Inhibitor (DBI) bindet an der Benzodiazepin-Bindestelle und verdrängt dadurch Benzodiazepine von dieser. Das Vorkommen von DBI im Nervensystem zeigt, dass es auch physiologische Agonisten der Benzodiazepin-Bindungsstelle gibt. Die Funktion dieses Peptids ist bisher unklar.
- Einige β-Carbolin-3-carboxamide wirken an der Benzodiazepin-Bindestelle als negative allosterische Modulatoren,[3] was bedeutet, dass sie die verschlossene Konformation des Ionenkanals begünstigen. Das Derivat FG-7142 bewirkt Angstzustände. Diese Wirkung wird jedoch, zumindest zum Teil, durch den 5-HT2C-Rezeptor vermittelt.[4] Dessen Aktivierung bewirkt GABA-erge Stimulation.[5]
- Flumazenil, ein kompetitiver Antagonist an der Benzodiazepin-Bindungsstelle, dient als Antidot der Benzodiazepin-Sedativa.[6]
- Die Allgemeinanästhetika Etomidat und Propofol wirken praktisch ausschließlich über GABAA-Rezeptoren. Sie vermitteln die immobilisierende Wirkung (keine Reaktion auf schmerzhafte Stimuli wie z. B. während eines chirurgischen Eingriffs) über GABAA-Rezeptoren im Rückenmark, die wahrscheinlich aus α3- und α5-, β3- und γ2-Untereinheiten zusammengesetzt sind. Die hypnotische (schlafinduzierende) Wirkung wird unter anderem von α5, β3 und γ2 enthaltenden GABAA-Rezeptoren vermittelt. Die genaue Lokalisation dieser Rezeptoren und anderer Untereinheiten, die Hypnose vermitteln, sind noch unbekannt. Die sedierende (keine Reaktion auf verbale Stimuli/Befehle) Wirkung von Etomidat und Propofol wird von α1, β2 und γ2 enthaltenden GABAA-Rezeptoren wahrscheinlich im Neocortex vermittelt.[7]
- 4-Hydroxybutansäure (GHB) spricht α4βδ GABAA-Rezeptoren an.[8]
Eine beträchtliche Anzahl von Substanzen, die stimulierend auf den GABAA-Rezeptor wirken, sind suchtauslösend. Dies gilt vor allem für die großen Substanzklassen der Barbiturate und Benzodiazepine. Vermittelt wird die Sucht über α1-haltige Rezeptoren auf Interneuronen im ventralen Tegmentum. Deren Reizung bewirkt in einem neuroplastischen Prozess die funktionelle Veränderung von AMPA-Rezeptoren, die sich auf diesem Neuron befinden.[9]
GABA-Rezeptoren unterliegen der pharmakologischen Toleranz. Das bedeutet, dass bei Gabe von Medikamenten, die den Rezeptor beeinflussen, mit der Zeit eine immer größere Dosis benötigt wird, um den gleichen Effekt zu erzielen.
Auslöser paradoxer Reaktionen
Eine Vielzahl von Substanzen, die im Normalfall als Agonisten auf den GABAA-Rezeptor wirken, können durch eine Abweichung in der Zusammenstellung der fünf Untereinheiten des Rezeptors (siehe Abbildung) zu einem Antagonisten werden. Sie erzielen damit eine gegenteilige Wirkung, die Paradoxe Reaktion genannt wird. Alkohol, zum Beispiel, wirkt dann nicht beruhigend oder entspannend, sondern erzeugt Nervosität und innere Unruhe.
Man geht davon aus, dass genetische und auch epigenetische Abweichungen den entsprechenden Veränderungen der GABAA Rezeptoren zugrunde liegen. Letztere werden als mögliche Ursache dafür angesehen, dass ein solcher struktureller Umbau der Rezeptoren erst im Laufe des Lebens auftritt, ausgelöst zum Beispiel durch besondere Belastungen.
Die paradoxen Reaktionen haben eine oft unterschätzte Bedeutung im Bereich der Anästhesiologie, der Behandlung von Epilepsie und in der Psychiatrie. Ist für eine hier eingesetzte Substanz die Möglichkeit einer paradoxen Reaktion bekannt, so wird empfohlen, den Behandlungsverlauf gezielt hierauf zu kontrollieren und pharmazeutische Gegenmittel (Antidot) bereitzuhalten.[10][11][12][13]
GABAA-rho-Rezeptoren
GABAA-ρ-Rezeptoren, früher als GABAC-Rezeptoren bezeichnet, werden nicht durch Bicucullin gehemmt und sind nahezu insensitiv gegen Benzodiazepine, Barbiturate und Neurosteroide. Sie aktivieren und inaktivieren langsamer als die GABAA-Rezeptoren und desensitivieren kaum.
GABAA-ρ-Rezeptoren kommen hauptsächlich in der Netzhaut und in geringerer Konzentration im Hippokampus, in den Colliculi superiores sowie im Rückenmark vor.
GABAA-ρ-Rezeptoren haben dieselbe Struktur wie GABAA-Rezeptoren und werden deshalb von der International Union of Pharmacology nicht als eigenständiger GABA-Rezeptor-Subtyp vorgeschlagen.[14] Unterschiede zwischen GABAA-ρ-Rezeptoren und GABAA-Rezeptoren sind ihre Pharmakologie (siehe oben), ihre Zusammensetzung (GABAA-ρ-Rezeptoren sind ausschließlich aus ρ-Untereinheiten aufgebaut, der typische GABAA-Rezeptor besteht aus α-, β- und γ-Untereinheiten) und die Zeitkonstanten ihrer Aktivierung und Deaktivierung. Es gibt die drei Untereinheiten ρ1–ρ3.
GABAB-Rezeptoren
Der metabotrope GABAB-Rezeptor ist ein G-Protein-gekoppelter Rezeptor und wird von einem Transmembranprotein mit sieben Transmembrandomänen gebildet. Die Signaltransduktion wird durch ein Gi-Protein vermittelt und führt postsynaptisch zu einer Aktivierung von ligandengesteuerten Kalium-Kanälen und so zu einem IPSP und präsynaptisch neben der Kalium-Kanal-Aktivierung auch noch zu einem Verschließen von spannungsgesteuerten Calciumkanälen. Häufig sind GABAB-Rezeptoren auch in der präsynaptischen Membran lokalisiert. Hier führt die Abnahme der Calcium-Konzentration zu einer reduzierten Transmitterfreisetzung aus der Präsynapse.[15] Agonist am GABAB-Rezeptor ist Baclofen. Funktionelle GABAB-Rezeptoren werden durch die heteromere Verbindung von GABAB1 mit GABAB2-Untereinheiten gebildet, welche zusammen einen Heterodimer bilden. Der Carboxy-Terminus von GABAB2 wiederum ist intrazellulär mit je zwei Tetrameren der sogenannten KCTD-Proteine ("KCTD" für potassium channel tetramerization domain-containing) verbunden. Die KCTD-Proteine verstärken die Antwort auf Agonisten, beschleunigen die G-Protein-Aktivierung und -Desensitisierung.[16]
Weiterführende Literatur
- Q. Wang, Y. Han, H. Xue: Ligands of the GABAA Receptor Benzodiazepine Binding Site. (PDF; 285 kB). In: CNS Drug Reviews. Band 5, 1999, S. 125.
- U. Rudolph, F. Knoflach: Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. In: Nature reviews. Drug discovery. Band 10, Nummer 9, Juli 2011, S. 685–697, doi:10.1038/nrd3502, PMID 21799515, PMC 3375401 (freier Volltext) (Review).
Quellen
- Susanne Rösner: Meta-Analyse zur Wirksamkeit von Acamprosat und Naltrexon in der Entwöhnungsbehandlung alkoholabhängiger Patienten. (PDF) 2006, abgerufen am 15. Dezember 2014.
- K. M. Höld u. a.: Alpha-thujone (the active component of absinthe): gamma-aminobutyric acid type A receptor modulation and metabolic detoxification. In: Proc. Natl. Acad. Sci. U.S.A. Band 97, 2000, S. 3826–3831. PMID 10725394 HTML
- J. Auta, P. J. Winsauer, W. B. Faust, P. Lambert, J. M. Moerschbaecher: Effects of negative allosteric modulators of gamma-aminobutyric acidA receptors on complex behavioral processes in monkeys. In: J Pharmacol Exp Ther. Band 280, Nr. 1, 1997, S. 316–325, PMID 8996212.
- E. A. Hackler, G. H. Turner, P. J. Gresch u. a.: 5-Hydroxytryptamine2C receptor contribution to m-chlorophenylpiperazine and N-methyl-beta-carboline-3-carboxamide-induced anxiety-like behavior and limbic brain activation. In: J. Pharmacol. Exp. Ther. Band 320, Nr. 3, 2007, S. 1023–1029, doi:10.1124/jpet.106.113357, PMID 17138863.
- R. W. Invernizzi, M. Pierucci, E. Calcagno u. a.: Selective activation of 5-HT(2C) receptors stimulates GABA-ergic function in the rat substantia nigra pars reticulata: a combined in vivo electrophysiological and neurochemical study. In: Neuroscience. Band 144, Nr. 4, 2007, S. 1523–1535, doi:10.1016/j.neuroscience.2006.11.004, PMID 17161544.
- Klaus Aktories, Ulrich Förstermann, Franz Hofmann, Klaus Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage. Elsevier, Urban & Fischer, München/ Jena 2009, ISBN 978-3-437-42522-6.
- U. Rudolph, B. Antkowiak: Molecular and neuronal substrates for general anaesthetics. In: Nat Rev Neurosci. Band 5, 2004, S. 709–720. PMID 15322529
- N. Absalom u. a.: α4βδ GABAA receptors are high-affinity targets for γ-hydroxybutyric acid (GHB). In: Proc Natl Acad Sci. Band 109, 2012, S. 13404. PMID 22753476
- C. Bocklisch, K. Tan, C. Lüscher: Warum Schlafmittel wie Valium süchtig machen. In: Spektrum der Wissenschaft. 2010. Artikelseite
- C. Robin, N. Trieger: Paradoxical reactions to benzodiazepines in intravenous sedation: a report of 2 cases and review of the literature. In: Anesthesia progress. Band 49, Nummer 4, 2002, S. 128–132, PMID 12779114, PMC 2007411 (freier Volltext) (Review).
- Carol Paton: Benzodiazepines and disinhibition: a review. In: Psychiatric Bulletin. 26, 2002, S. 460, doi:10.1192/pb.26.12.460, PDF.
- T. Bäckström, M. Bixo, M. Johansson, S. Nyberg, L. Ossewaarde, G. Ragagnin, I. Savic, J. Strömberg, E. Timby, F. van Broekhoven, G. van Wingen: Allopregnanolone and mood disorders. In: Progress in neurobiology. Band 113, Februar 2014, S. 88–94, doi:10.1016/j.pneurobio.2013.07.005, PMID 23978486 (Review), PDF.
- E. N. Brown, R. Lydic, N. D. Schiff: General anesthesia, sleep, and coma. In: The New England Journal of Medicine. Band 363, Nummer 27, Dezember 2010, S. 2638–2650, doi:10.1056/NEJMra0808281, PMID 21190458, PMC 3162622 (freier Volltext) (Review).
- E. A. Barnard u. a.: International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. In: Pharmacol Rev. Band 50, 1998, S. 291–313. PMID 9647870 HTML
- K. Aktories: Pharmakologie und Toxikologie. 9. Auflage.
- J. Schwenk, M. Metz, G. Zolles u. a.: Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. In: Nature. Band 465, Nr. 7295, 2010, S. 231–235, doi:10.1038/nature08964, PMID 20400944.