Homologie (Genetik)
Zwei Gene (oder Proteine) sind zueinander homolog, wenn sie von einem gemeinsamen Vorläufer abstammen.
Stimmen zwei Gene in der Nukleotidsequenz in mehr als 30 % ihrer Nukleotide in der Abfolge überein, so gilt eine andere Ursache als die gemeinsame Abstammung als unwahrscheinlich; man betrachtet diese beiden Gene daher als homolog. Ähnliches gilt für die Aminosäuresequenz der aus den Genen produzierten Proteine, wobei die Homologie bei einer Sequenzübereinstimmung von über 10 % angenommen wird.
Homologie von Genen führt nicht zwangsläufig zu Homologie von Organen, so ist z. B. der Fall nicht auszuschließen, dass homologe Gene und damit auch homologe Proteine in zwei völlig verschiedenen, nicht-homologen Geweben gefunden werden.[1]
Homologe Chromosomen enthalten in der gleichen Reihenfolge der Genorte die gleichen Gene, doch können diese als verschiedene Allele vorliegen. Damit können sich in einer diploiden Zelle die Chromosomen väterlicher bzw. mütterlicher Herkunft unterscheiden.
Dieser Homologiebegriff ist zu unterscheiden von der Homologie verschiedener Organe, die in der Phylogenetik und Evolutionstheorie betrachtet wird.
Homologie zwischen Genen verschiedener Arten
Homologie zwischen zwei Genen kann nur dann festgestellt werden, wenn sich die Sequenzen noch nicht so weit voneinander entfernt haben, dass die Ähnlichkeit untereinander nur noch so groß wie zwischen zwei zufälligen Sequenzen ist. Dies hängt nicht nur von der abgelaufenen Zeit ab, sondern auch vom Grad der Konservierung der jeweiligen Sequenz. Die Enzyme wichtiger Stoffwechselwege wie der Glykolyse sind in hohem Maß konserviert:
| Protein im Mensch | Identität zum Protein im Organismus:[2] | |||||
|---|---|---|---|---|---|---|
| Schimpanse (Vorläufer 5–6 Mio. J.) |
Hausratte (Vorläufer 100–150 Mio. J.) |
Zebrafisch (Vorläufer 200–300 Mio. J.) |
Lanzett- Fischchen (Vorl. 500 Mio. J.) |
Fadenwurm (Vorläufer 800–1000 Mio. J.) |
Escherichia coli | |
| PFK | 100 % | 94 % | 77 % | 63 % | 40 % | 40 % |
| α-Hämoglobin | 100 % | 78 % | 53 % | 31 % | kein Ortholog | kein Ortholog |
| Insulin | 98 % | 82 % | 46 % | kein Ortholog | kein Ortholog | kein Ortholog |
| EPO | ±67 % | 80 % | 36 % | kein Ortholog | kein Ortholog | kein Ortholog |
Eine Aminosäuresequenz-Identität von 60 bis 80 % ist beispielsweise üblich zwischen homologen Säugetiergenen, eine solche von 40 bis 60 % zwischen homologen Wirbeltiergenen. Sinkt die Identität unter 10 % (5 % entsprechen einem zufälligen Ergebnis), wäre eine ggf. vielleicht bestehende Homologie tatsächlich nicht mehr nachweisbar. Tatsächlich ist bereits unterhalb 30 % das Vorliegen einer Homologie fraglich, da unabhängige Proteindomänen nicht selten eine gewisse Ähnlichkeit besitzen (Parallelevolution). Weitere Ausnahmen bilden kurze und Tandem-Sequenzen, da hier die Wahrscheinlichkeit einer zufälligen Ähnlichkeit größer ist.
Homologie zwischen verdoppelten oder fremden Genen
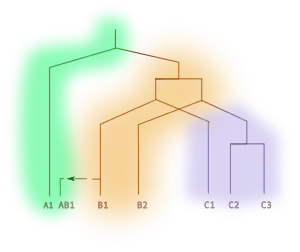
Gäbe es nur Artneubildungsereignisse, dann gäbe es für jedes Gen ein entsprechendes Gen in jedem anderen Organismus. Genverdopplung und horizontaler Gentransfer können zu zusätzlichen Genkopien führen, die sich im Lauf der Zeit mit der Gendrift voneinander weg entwickeln. Zur klareren Unterscheidung der Homologieverhältnisse zwischen solchen Genen werden die folgenden Bezeichnungen verwendet: Zwei Gene sind zueinander paralog, wenn ihr gemeinsames Vorläufergen eine Genverdopplung durchlaufen hat. Zwei Gene sind zueinander ortholog, wenn ihr gemeinsamer Urahn ein Artbildungsereignis durchlaufen hat. Sie sind xenolog, wenn eines von beiden einen horizontal transferierten Vorfahren hat (siehe Abbildung, B1 und C1 sind zueinander ortholog, B1 und B2 paralog, A1 und AB1 xenolog).[3]
Weiterhin spricht man von (1:n)-Orthologie, wenn es sich um Sequenz X in Spezies A und Sequenz Y in Spezies B handelt und es eine zu Y paraloge Sequenz Z in Spezies B gibt, die zu X direkt ortholog ist. Umgekehrt sind Y und X (n:1)-Orthologe. In diesen beiden Fällen hat also in einer der Spezies eine Diversifizierung stattgefunden und zwischen X und Y liegt eine Genverdopplung (siehe Abbildung, B1 und C2). Schließlich noch bedeutet (m:n)-Orthologie zwischen X und Y, dass in beiden Spezies eine Diversifizierung stattgefunden hat und daher zwei Genverdopplungen zwischen X und Y liegen (in der Abbildung nicht dargestellt). Entsprechend wird in diesem Zusammenhang strikte Orthologie als (1:1)-Orthologie bezeichnet. Reale Beispiele für diese Beziehungen sind die Verhältnisse zwischen einzelnen MADS-Box-Proteinen.[4]
Genverdopplung und horizontaler Gentransfer sind die häufigsten biologischen Prozesse, bei denen die Anzahl der Gene erhöht wird. Eine Verringerung der Anzahl der Gene geschieht durch Deletion oder Insertion von Nonsense-Sequenzen.
Genetischer Abstand
Im Vergleich der Basensequenz homologer Gene können Grade der Übereinstimmung und damit genetische Abstände verwandter Arten ermittelt werden. Ist zudem ein Zeitpunkt des gemeinsamen letzten Vorfahren dieser beiden Arten bekannt, lassen sich aus dem Grad der Übereinstimmung dieser Gene auch für weitere nahe verwandte Arten die Zeitpunkte ihrer letzten gemeinsamen (noch unbekannten) Vorfahren ermitteln.[5][6]
Der Grad der genetischen Übereinstimmung kann im Zuge eines Sequenzalignment unterschiedlich betrachtet werden,[7] wodurch sich bei einer molekularen Uhr auch Diskrepanzen in der Rekonstruktion des zeitlichen Verlaufs verschiedener Untersucher ergeben können:
- Der Fixierungsindex ist vergleichsweise einfach anwendbar und daher ein häufig gebrauchtes relatives Maß mit Werten zwischen 0 (einer Art angehörend) und 1 (unterschiedlichen Arten angehörend).[8][9]
- Nei's Standard des genetischen Abstandes bezieht sich auf Punktmutationen und genetische Drift.[10]
- Cavalli-Sforza und Edwards 1967 bezieht sich auf genetische Drift.[11]
- Reynolds, Weir und Cockerham's 1983 bezieht sich auf genetische Drift.[12]
- Nei's DA Abstand bezieht sich auf Punktmutationen und genetische Drift und gibt besonders zuverlässige Verwandtschaftsbeziehungen, auch auf Basis von Satelliten-DNA.[13]
Proteom
Eine Variante dieser Homologie der Gene ist die Homologie der kodierten Proteine, d, h. innerhalb des Proteoms statt des Genoms. Wegen der Degeneration des Genetischen Codes (mehrere Basentripletts kodieren dieselbe Aminosäure) bleiben hierbei Auswirkungen der Gendrift (siehe auch Molekulare Uhr) außen vor. Ein Beispiel für Homologie-Analysen auf Proteinebene findet sich etwa bei Adriaenssens, Krupovic et al. (2020)[14]
Literatur
- G. S. Gray, W. M. Fitch: Evolution of antibiotic resistance genes: the DNA sequence of a kanamycin resistance gene from Staphylococcus aureus. In: Molecular biology and evolution. Band 1, Nummer 1, Dezember 1983, S. 57–66, ISSN 0737-4038. PMID 6100986.
- R. A. Jensen: Orthologs and paralogs - we need to get it right. In: Genome biology. Band 2, Nummer 8, 2001, S. INTERACTIONS1002, ISSN 1465-6914. PMID 11532207. PMC 138949 (freier Volltext).
Einzelnachweise
- Werner A. Müller, Monika Hassel: Entwicklungsbiologie. 2006, ISBN 3-540-24057-8, S. 252. (online)
- UniProt
- W. M. Fitch: Homology. A personal view on some of the problems. In: Trends in Genetics. Band 16, Nr. 5, Mai 2000, S. 227–231 (228), PMID 10782117 (Online [PDF; 115 kB]).
- Klaus D. Grasser: Annual Plant Reviews, Regulation of Transcription in Plants. Volume 29. Wiley-Blackwell, 2006, ISBN 1-4051-4528-5, S. 37.
- Masatoshi Nei, A. K. Roychoudhury: Sampling variances of heterozygosity and genetic distance. In: Genetics. Band 76, Nr. 2, 1974, S. 379–390. genetics.org
- Rike Stelkens, Ole Seehausen: Genetic distance between species predicts novel trait expression in their hybrids. In: Evolution. Band 63, Nr. 4, 2009, S. 884–897. doi:10.1111/j.1558-5646.2008.00599.x
- Population Genetics IV: Genetic distances -- biological vs. geometric approaches.
- Masatoshi Nei, Ronald K. Chesser: Estimation of fixation indices and gene diversities. In: Annals of Human Genetics. Band 47, Nr. 3, 1983, S. 253–259. doi:10.1111/j.1469-1809.1983.tb00993.x
- Jérôme Goudet: FSTAT (version 1.2): a computer program to calculate F-statistics. In: Journal of heredity. Band 86, Nr. 6, 1995, S. 485–486. jhered.oxfordjournals.org
- M. Nei: Genetic distance between populations. In: Am. Nat. Band 106, 1972, S. 283–292.
- L. L. Cavalli-Sforza, A. W. Edwards: Phylogenetic analysis. Models and estimation procedures. In: American Journal of Human Genetics. Band 19, Nummer 3 Pt 1, Mai 1967, S. 233–257. PMID 6026583, PMC 1706274 (freier Volltext).
- John Reynolds, Bruce S. Weir, C. Clark Cockerham: Estimation of the coancestry coefficient: basis for a short-term genetic distance. In: Genetics. Band 105, Nr. 3, 1983, S. 767–779. genetics.org
- N. Takezaki, Masatoshi Nei: Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. In: Genetics. Band 144, 1996, S. 389–399.
- Evelien M. Adriaenssens, Mart Krupovic et al.: Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee, in: Archives of Virology 165, 11. März 2020, S. 1253–1260, doi:10.1007/s00705-020-04577-8, PDF, siehe dort unter §Chaseviridae