Günter Schultz
Günter Schultz (* 23. Januar 1936 in Frankfurt am Main; † 14. August 2021[1]) war ein deutscher Pharmakologe. Er hatte wesentliche Erkenntnisse über die biologische Signalverarbeitung gewonnen. Außerdem hatte er zahlreiche Jüngere zu diesem Forschungsgebiet hingeführt und es so vor allem in Deutschland mitgeprägt.

Leben
Seine Eltern waren der Arzt Ernst-Gottfried Schultz und dessen Ehefrau Margarete geb. Eichner. Nach dem Besuch des Charlottenburger Gymnasiums und dem Abitur 1955 studierte er an der Freien Universität Berlin Medizin. 1963 erhielt er die Approbation als Arzt und wurde mit einer Dissertation „Untersuchungen über das morphologische und sekretorische Verhalten der Corpusschleimhaut des Magens bei chronischer Hepatitis und Lebercirrhose“ zum Dr. med. promoviert. Er trat in die Arbeitsgruppe von Gerhard Senft (1926–1967) an dem von Hans Herken geleiteten Pharmakologischen Institut der Freien Universität ein. Nach Senfts Tod wechselte er 1968 an das Pharmakologische Institut der Ruprecht-Karls-Universität Heidelberg, dessen Leitung im selben Jahr Franz Gross übernahm. In Heidelberg habilitierte er sich 1970 für Pharmakologie mit einer Arbeit „Wirkungen von Hormonen und Pharmaka auf den Stoffwechsel cyclischer Nucleosid-3'-5'-monophosphate in der Rattenniere“. 1970 heiratete er auch die medizinisch-technische Assistentin Karin Munske (1941–2009), Mitarbeiterin in seiner Gruppe und Ko-Autorin einiger Publikationen erst unter ihrem Mädchennamen, dann als Karin Schultz. Von 1971 bis 1973 arbeitete er als Visiting Scientist (Gastwissenschaftler) bei Earl Wilbur Sutherland in der Gruppe von Joel Griffith Hardman (* 1933) an der Physiologischen Abteilung der Vanderbilt University in Nashville, Tennessee. Sutherland hatte um 1960 das cyclische Adenosinmonophosphat (cAMP) als einen second messenger bei der Wirkung chemischer Botenstoffe wie des Adrenalins sowie das cAMP-bildende Enzym Adenylylcyclase und das cAMP-spaltende Enzym Phosphodiesterase entdeckt und dafür 1971, im Jahr als Schultz in sein Labor eintrat, den Nobelpreis für Physiologie oder Medizin erhalten. Von 1973 bis 1983 bekleidete Schultz eine C3-Professur in Heidelberg, verbrachte aber wieder einige Monate an der Vanderbilt University. 1983 wurde er als C4-Professor – ordentlicher Professor – an die Freie Universität Berlin berufen und folgte am 1. Oktober Hans Herken als Direktor des Pharmakologischen Instituts an der Thielallee in Dahlem. 2003 wurde er pensioniert. Die Übergabe des Lehrstuhls an seinen Nachfolger Walter Rosenthal, den Leiter des Berliner Forschungsinstituts für Molekulare Pharmakologie, späteren Leibniz-Instituts für Molekulare Pharmakologie, fiel in die schwierige Zeit der Zusammenlegung der Medizinischen Fakultäten der Freien Universität und der Humboldt-Universität.
Forschung
Diuretika, cyclisches AMP und die Phosphodiesterase
Senft interessierte sich für Diuretika. Manche, besonders die Thiaziddiuretika sowie das chemisch verwandte, allerdings nicht diuretisch wirkende Diazoxid, erhöhten den Blutzuckerspiegel. Die Berliner Gruppe erwog, dass dabei cAMP eine Rolle spielen könnte, das den Kohlenhydratstoffwechsel regelt. Schultz baute eine Methode zur Messung von cAMP auf.[2] In der Tat hemmten sowohl Diazoxid als auch die Thiaziddiuretika die cAMP-abbauende Phosphodiesterase.[3][4] Vermutlich ist Phosphodiesterase-Hemmung nicht die Hauptursache für die Blutzuckerwirkung der Thiazide und des Diazoxids. Schultz aber hatte mit diesen Arbeiten zu seinem Thema gefunden, der biologischen Signalverarbeitung.
Cyclisches GMP und die Guanylylcyclase
Kurz bevor er Berlin verließ, war ein zweites cyclisches Nucleotid entdeckt worden, das cyclische Guanosinmonophosphat (cGMP). Ihm wandte sich Schultz in Heidelberg zuerst zu. „In den letzten Jahren wurde die Ausscheidung von cyclischem Guanosin-3',5'-monophosphat [..] im Urin von Ratten beschrieben […]. Über Bildung und Vorkommen dieses cyclischen Nucleotids in tierischen Geweben ist bisher nichts bekannt.“[5] Schultz und seine Mitarbeiter, darunter sein erster Doktorand Eycke Böhme (1943–1993), entdeckten „Bildung und Vorkommen“ – und damit die cGMP-bildende Guanylylcyclase, genauer die im Zytosol gelöste Guanylylcyclase – gleichzeitig mit zwei US-amerikanischen Gruppen, darunter Hardman und Sutherland, in vielen Organen.[5][6] Die Guanylylcyclase ist das Gegenstück zur cAMP-bildenden Adenylylcyclase. Sie existiert in mehreren Isoformen.[7][8] An deren Reinigung und Charakterisierung[9][10] bis zur Strukturaufklärung durch Klonierung ihrer Gene[11][12] hat die Gruppe in Heidelberg und später Berlin weiter mitgewirkt.
Auf der Suche nach der Funktion von cGMP fand Schultz in Nashville, dass im Samenleiter von Ratten Noradrenalin, Acetylcholin und Carbachol, die das Organ zur Kontraktion bringen, den Gehalt an cGMP steigerten.[13][14] cGMP war aber keineswegs ein second messenger für die Kontraktion – im Gegenteil. Das wurde 1977 klar. Wie Schultz' Gruppe und die Gruppe von Ferid Murad in den USA fanden, erhöhten auch mehrere Stoffe, die die glatte Muskulatur erschlaffen lassen, insbesondere die Nitrovasodilatatoren, den Gehalt an cGMP.[15] Außerdem bewirkte ein cGMP-Derivat selbst Erschlaffung.[16] cGMP war also ein second messenger nicht für Kontraktion, sondern für Erschlaffung. Ab 1980 wurde mit der Erkennung von Stickstoffmonoxid als dem körpereigenen Aktivator der zytosolischen Guanylylcyclase – anders ausgedrückt mit der Erkennung der zytosolischen Guanylylcyclase als des Rezeptors für körpereigenes Stickstoffmonoxid – die große physiologische Bedeutung des Enzyms klar.
Hemmung der Adenylylcyclase
Adrenalin löst seine Wirkungen über zwei Typen von Rezeptoren aus, die α- und β-Adrenozeptoren. Sutherlands Stimulation der Adenylylcyclase geschieht über β-Adrenozeptoren. Was die α-Adrenozeptoren angeht, so hatte man um 1970 beobachtet, dass ihre Aktivierung den cAMP-Gehalt von Organen verminderte. An Membranen von Thrombozyten (Blutplättchen) erforschten Schultz und sein zweiter Heidelberger Doktorand Karl-Heinrich Jakobs (* 1941) den Mechanismus. Er bestand in einer Hemmung der Adenylylcyclase.[17] Es gab also eine gegenläufige biologische Signalverarbeitung über das Enzym, Stimulierung und Hemmung. Über β-Adrenozeptoren und Stimulierung der Adenylylcyclase hemmte Adrenalin die Thrombozytenaggregation, über α-Adrenozeptoren und Hemmung der Adenylylcyclase dagegen förderte es die Thrombozytenaggregation. Die α-Adrenozeptoren unterschieden sich von jenen, die Glattmuskelkontraktion auslösen,[18] und wurden 1979 als α2-Adrenozeptoren identifiziert.[19]
Gs, Gi und darüber hinaus
Ebenfalls seit etwa 1970 war bekannt, dass Adrenalin und andere Botenstoffe wie Glukagon die Adenylylcyclase nur in Gegenwart von Guanosintriphosphat (GTP) stimulierten, wobei GTP in Guanosindiphosphat (GDP) und Phosphationen gespalten wurde. GTP trat dazu nicht mit dem Rezeptor oder der Adenylylcyclase in Kontakt, sondern mit einem separaten Proteinkomplex aus drei verschiedenen Untereinheiten α, β und γ, einem heterotrimeren G-Protein (GTP-bindenden Protein). Auch die Hemmung der Adenylylcyclase in Blutplättchen durch Adrenalin erwies sich als GTP-abhängig.[20] Außer über α2-Adrenozeptoren wird die Adenylylcyclase über manche Muscarinrezeptoren, Rezeptoren für den Neurotransmitter Acetylcholin, gehemmt, zum Beispiel im Herzen. Wiederum erforderte die Hemmung die Anwesenheit von GTP.[21] Die Frage der G-Protein-Forschung um 1980 lautete „whether the same, or distinct, G-proteins confer activating and attenuating signals to the adenylate cyclase enzyme“, ob es ein und dasselbe G-Protein ist oder verschiedene G-Proteine sind, die einerseits stimulierende, andererseits hemmende Signale an die Adenylylcyclase weitergeben.[22]
Zwei Heidelberger Experimente von Schultz, Jakobs und einem Post-Doktoranden, Klaus Aktories, entschieden 1983 für verschiedene G-Proteine. Das erste wurde an Lymphomzellen durchgeführt, deren Adenylylcyclase eines Gendefekts wegen auf stimulierende Botenstoffe nicht mehr reagierte. Der Adenylylcyclase-hemmende Botenstoff Somatostatin dagegen verursachte bei diesen Zellen die übliche Hemmung: Es fehlte des Gendefekts wegen das stimulierende G-Protein Gs – „s“ für – „stimulierend“, nicht aber das davon verschiedene inhibierende G-Protein Gi – „i“ für „inhibierend“.[23] Das zweite Experiment wurde an Blutplättchen durchgeführt, deren Adenylylcyclase, wie oben erwähnt, durch Adrenalin über α2-Adrenozeptoren gehemmt wird. Das Prostaglandin Prostaglandin-E1 stimuliert die Blutplättchen-Adenylylcyclase. Ein bakterielles Toxin, das vom Erreger des Keuchhustens stammende Pertussistoxin, verhinderte die Hemmung durch Adrenalin und die begleitende Spaltung von GTP zu GDP und Phosphationen, nicht aber die Stimulierung durch Prostaglandin-E1:[24] Pertussistoxin blockierte Gi, nicht aber das davon verschiedene Gs.
Die G-Proteine wurden weltweit eines der intensivst beforschten biologischen Themen. 1983 kannte man drei, Gs, Gi und das in den Photorezeptorzellen der Netzhaut der Lichtwahrnehmung dienende Transducin. 1984 kam ein weiteres hinzu, das wie Gi durch Pertussistoxin blockiert wurde, Go, in dessen Bezeichnung „o“ für „other“ steht, „ein anderes“ G-Protein. Vor allem durch Klonierung der Gene wurde die Vielfalt immer größer. 2005 waren 17 verschiedene Gene für α-, 5 verschiedene Gene für β- und 12 verschiedene Gene für γ-Untereinheiten bekannt, aus denen durch alternatives Spleißen eine noch größere Zahl von Proteinen resultierte.[25] Die aus ihnen zusammengesetzten Heterotrimere teilt man nach den α-Untereinheiten in vier Klassen ein, Gs, Gi/o, Gq/11 und G12/13. Über fast tausend „G-Protein-gekoppelte Rezeptoren“ können Botenstoffe, auch viele Arzneistoffe, auf die G-Proteine und dadurch weiter auf „Effektoren“, also Enzyme oder Ionenkanäle wirken.[26]
Gi, Go und die Modulation von Calciumkanälen
Schultz zeigte 1987 und 1988 in Berlin zusammen mit Walter Rosenthal und den Homburger Elektrophysiologen Wolfgang Trautwein und Jürgen Hescheler (* 1959) erstmals eine Beeinflussung spannungsgesteuerter Calciumkanälen durch G-Proteine, zunächst bei Nervenzellen. Ein Opioid hemmte den Eintritt von Calciumionen in die Zellen. Die Hemmung wurde durch Pertussistoxin blockiert. Wurde ein Gemisch von Gi und Go in die Pertussistoxin-behandelten Zellen injiziert, so trat die Hemmung durch das Opioid wieder auf: Die Hemmung wurde durch das injizierte Gi-Go-Gemisch „rekonstituiert“.[27] Die Veröffentlichung erregte sogleich Aufsehen,[28] zumal das verantwortliche G-Protein anscheinend Go war, dem damit erstmals eine Rolle bei der Signalverarbeitung zugeschrieben werden konnte.
Die gegenteilige Rolle spielten G-Proteine in der Nebennierenrinde. In deren Zellen steigerte Angiotensin II den Calciumeintritt durch spannungsabhängige Calciumkanäle, und Pertussistoxin blockierte die Steigerung. Immunologisch ließ sich Go ausschließen und ein Gi-Protein identifizieren.[29] In einer weiteren endokrinen Drüse schließlich, dem Hypophysenvorderlappen – „GH3“-Zellen aus einem Hypophysenvorderlappentumor von Ratten – wurde der Calciumeintritt durch Somatostatin gehemmt und durch LHRH – das Luteinisierungshormon-freisetzende Hormon – gesteigert. Pertussistoxin blockierte die Hemmung wie die Steigerung. Hemmung und Steigerung wurden anscheinend durch verschiedene G-Proteine vermittelt, denn die Zellen enthielten sowohl Go als auch ein Gi-Protein.[30]
Selektivität und Komplexität
Aus der unübersehbaren Zahl von G-Protein-Untereinheiten und G-Protein-Heterotrimeren wählen Zellen bestimmte, für ihre jeweilige Funktion adäquate Kombinationen. Das wurde zuerst durch eine „surprising series of experiments“,[31] „eine staunenerregende Versuchsserie“ an GH3-Hypophysenvorderlappenzellen deutlich. Deren Calciumkanäle werden außer, wie oben erwähnt, über Somatostatin-Rezeptoren auch über Muscarinrezeptoren gehemmt. Schultz injizierte mit dem Molekularbiologen Burghardt Wittig, ihrer beider Doktorandin Christiane Kleuss und anderen in die Zellkerne von GH3-Zellen Antisense-Oligonukleotide zur präzisen Unterdrückung der Synthese bestimmter G-Protein-Untereinheiten, zunächst α-,[32] dann β-,[33] schließlich γ-Untereinheiten[34] und behandelte die Zellen dann mit Somatostatin oder Carbachol. Die Schlussfolgerung in der dritten Publikation:[34] „Bei jedem der beiden Signalübersetzungswege ist anscheinend eine spezifische γ-Untereinheit mit einer spezifischen β-Untereinheit und einer spezifischen αo-Untereinheit komplexiert, so dass zwei spezifische G-Proteine resultieren. … Der Muscarinrezeptor ist an ein aus αo1/β3/γ4 bestehendes, der Somatostatin-Rezeptor an ein aus αo2/β1/γ3 bestehendes G-Protein gekoppelt. Beide G-Proteine vermitteln eine Hemmung spannungsabhängiger Calciumkanäle.“
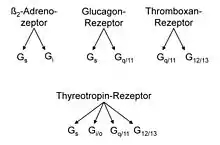
Mit derselben Methode untersuchte Schultz' Gruppe die Signalverarbeitung an den GH3-Rezeptoren für TRH – das Thyreotropin-freisetzende Hormon. TRH förderte wie LHRH (siehe oben) den Calciumeintritt. Die Wirkung, genauer der durch Pertussistoxin hemmbare Teil der Wirkung, wurde durch G-Proteine mit den α-Untereinheiten αi2 und αi3 vermittelt.[35]
Der Doktorand Karl-Ludwig Laugwitz (* 1968)[36] und der Post-Doktorand Stefan Offermanns entwickelten eine weitere Methode zur Analyse von G-Proteinen, Photoaffinitätsmarkierung gefolgt von immunologischer Identifizierung. Damit wurde die Signalverarbeitung an den Rezeptoren der Schilddrüse für das die Drüse stimulierende Thyreotropin untersucht. Effektoren sind dabei die Adenylylcyclase, die den second messenger cAMP bildet, und eine Phospholipase C, die die second messenger Inositoltrisphosphat und Diacylglycerin bildet. Beide Enzyme werden durch Thyreotropin stimuliert. G-Protein-Analyse ergab zunächst, dass daran die G-Protein-Klassen Gs und Gq/11,[37] im weiteren, dass auch die Klassen Gi/o und G12/13 beteiligt waren, mit insgesamt zehn verschiedenen α-Untereinheiten, ein, wie die Autoren schreiben, unerwarteter Befund, der zeige, dass neben spezifischen auch multiple Rezeptor-G-Protein-Kopplungen vorkommen.[38]
Spezifisch hingegen waren Signalkaskaden bei der Wirkung von Thromboxan A2 und Adenosindiphosphat auf Blutplättchen. Beide sind wichtige Mediatoren der Thrombocytenaggregation. Thromboxan A2 aktivierte neben anderen G-Proteinen G12 und G13; das war – 1994 – der erste Nachweis einer biologischen Funktion dieser beiden G-Proteine.[39] Spezifisch über G13 und die kleine GTPase Rho als Effektor löste Thromboxan A2 dann den mit der Aggregation einhergehenden Formwandel der Plättchen aus.[40][25] Adenosindiphosphat dagegen hemmte über seinen Rezeptor, den „Purinozeptor“ P2Y12, und Gi2 die Adenylylcyclase.[41]
Auch an der Kontraktion der glatten Muskulatur von Blutgefäßen durch Endothelin-1 waren G12/13 und als Effektor die kleine GTPase Rho beteiligt.[42]
Selektivität und Komplexität – „Diversity and Selectivity“: „Mit der Vorstellung einer linearen Signalübersetzung – ein Rezeptor koppelt sich an ein G-Protein, das dann einen Effektor aktiviert – lassen sich die experimentellen Befunde nicht erklären. … G-Protein-vermittelte Signalverarbeitung ist vielmehr ein komplexes Netzwerk mit Divergenzen und Konvergenzen bei jedem Schritt, vom Rezeptor zum G-Protein zum Effektor.“[43]
TRP-Kanäle
Schultz' jüngstes Forschungsgebiet, ab Beginn der 1990er Jahre, geht auf Mutageneseuntersuchungen an der Taufliege Drosophila melanogaster zurück. Eine von zahlreichen Mutationen hatte dazu geführt, dass die Photorezeptoren auf Belichtung mit einem zu kurzen elektrischen Strom antworteten, einem transient receptor potential; helles Licht blendete die Fliegen deshalb. 1985 bis 1989 hatte man als Ursache das Fehlen eines neuartigen Calciumkanals erkannt, eines TRP-Kanals, transient receptor potential channel, benannt nach der Fehlfunktion bei seiner Deletion.[44] Angesichts der Bedeutung von Calcium bei der biologischen Signalverarbeitung begann sogleich die Suche nach ähnlichen Ionenkanälen bei Säugern. 1995 berichteten zwei US-amerikanische Gruppen über die Klonierung eines dem Drosophila-trp-Gen homologen menschlichen Gens. Schultz' Gruppe folgte 1996, ging aber weiter, exprimierte das homologe Gen in tierischen Zellen (CHO-Zellen) und erhielt in der Tat einen Kationenkanal[45] – den ersten TRP-Kanal von Säugern, TRPC1.[46] Besonderes Interesse gewann er, weil die Autoren vermuteten, er könne jener Kanal in der Zellmembran sein, durch den Zellen ihre Calciumspeicher nach Entleerung wieder auffüllen, der CRAC-Kanal, calcium release actived calcium channel, oder speichergesteuerte Kanal, store-operated channel.
Nach 1995 bis 1996 proliferierte die TRP-Forschung rasch. Schultz' Gruppe hat erheblich zur Klonierung und Charakterisierung etlicher der heute (2013) bekannten 26 TRP-Proteine beigetragen. Zur Bildung der CRAC-Kanäle scheinen außer TRP-Kanälen der TRPC-Familie weitere Proteine beizutragen.[47] Kandidaten als chemische Signale für die Öffnung der CRAC-Kanäle sind die durch Phospholipase C gebildeten second messenger Inositoltrisphosphat und Diacylglycerin. In der Tat öffnete Diacylglycerin TRPC3- und TRPC6-Kanäle.[48] Aber auch ganz andere Reize, chemische wie physikalische, öffnen manche TRP-Kanäle, so Kälte und Hitze, das ein Wärmegefühl hervorrufende Capsaicin und das ein Kältegefühl hervorrufende Menthol. Der OTRPC4- oder TRPV4-Kanal, den Schultz Gruppe klonierte, reagierte auf Änderungen des osmotischen Drucks im Extrazellularraum.[49]
Im Jahr 2000 haben Schultz und Mitarbeiter die TRP-Kanäle in eine Ordnung mit drei Familien gebracht,[50] die mit Erweiterungen – sechs Familien – Bestand hat.[51][46]
Forschungsorganisation
Zusätzlich zu seiner Tätigkeit als Arbeitsgruppenleiter und Institutsdirektor hinaus widmete sich Schultz übergeordneten Aufgaben. 1988 gründete er den Forschungsschwerpunkt der Deutschen Forschungsgemeinschaft „Molekulare Mechanismen der Signaltransduktion in Membranen“, den er bis 1994 koordinierte. Aus dem Forschungsschwerpunkt ging 1994 der bis 2005 bestehende Sonderforschungsbereich 366 (SFB 366) „Zelluläre Signalerkennung und -umsetzung“ hervor, mit dem Biochemiker Werner Reutter als Sprecher und Schultz als stellvertretendem Sprecher. Zeitweilig gehörten ihm acht Teilprojekte aus dem Pharmakologischen Institut an. Im Rahmen der Dahlem-Konferenzen veranstaltete der SFB jährlich internationale Tagungen über „Signaltransduktion“. Von 1994 bis 2000 war Schultz im Senat der Deutschen Forschungsgemeinschaft. Er war lange Hauptherausgeber der Zeitschrift Molecular and Cellular Endocrinology und der Buchreihe Reviews of Physiology, Biochemistry and Pharmacology.
Schüler
Folgende Wissenschaftler haben sich in Schultz' Heidelberger Arbeitsgruppe oder seinem Berliner Institut habilitiert (Jahr der Habilitation):
- Franz Hofmann (1977), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Technischen Universität München und Gründungsdirektor des Berliner Forschungsinstituts für Molekulare Pharmakologie; Hofmanns Habilitation wurde von Franz Gross mitbetreut
- Eycke Böhme (1978), später Professor am Berliner Institut; auch seine Habilitation wurde von Franz Gross mitbetreut
- Karl Heinrich Jakobs (1979), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Universität-Gesamthochschule Essen
- Klaus Aktories (1983), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Universität des Saarlandes in Homburg und dann der Albert-Ludwigs-Universität Freiburg
- Walter Rosenthal (1990), später zu Schultz Nachfolger berufen und seit 2009 Direktor des Max-Delbrück-Centrums für Molekulare Medizin in Berlin-Buch
- Roland Seifert (1992), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Universität Regensburg und dann der Medizinischen Hochschule Hannover
- Doris Koesling (1996), später Lehrstuhlinhaberin für Pharmakologie und Toxikologie der Ruhr-Universität Bochum
- Bernd Nürnberg (1997), später Lehrstuhlinhaber für Biochemie und Molekularbiologie der Heinrich-Heine-Universität Düsseldorf und dann Lehrstuhlinhaber für Pharmakologie und Experimentelle Therapie der Eberhard Karls Universität Tübingen
- Thomas Gudermann (1998); später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Philipps-Universität Marburg und dann der Ludwig-Maximilians-Universität München
- Stefan Offermanns (1998), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Ruprecht-Karls-Universität Heidelberg und dann Direktor des Max-Planck-Instituts für Herz- und Lungenforschung in Bad Nauheim
- Torsten Schöneberg (2001), später Lehrstuhlinhaber für Molekulare Biochemie der Universität Leipzig
- Michael Schaefer (2001), später Lehrstuhlinhaber für Pharmakologie und Toxikologie der Universität Leipzig
- Christian Harteneck (2003), später Professor für Pharmakologie in Tübingen
Anerkennung
1994 erhielt Schultz den Max-Planck-Forschungspreis und 1999 den Feldberg-Preis für britisch-deutschen wissenschaftlichen Austausch. 2001 wurde er Mitglied der Deutschen Akademie der Naturforscher Leopoldina.[52] 2003 wurde er mit dem Bundesverdienstkreuz 1. Klasse ausgezeichnet.
Literatur
- Eberhard Hackenthal, Stefan Offermanns, Günter Schultz: Pharmakologisches Institut, Medizinische Fakultät der Ruprecht-Karls-Universität Heidelberg. In: Athineos Philippu (Hrsg.): Geschichte und Wirken der pharmakologischen, klinisch-pharmakologischen und toxikologischen Institute im deutschsprachigen Raum, S. 329–336. Berenkamp-Verlag, Innsbruck 2004. ISBN 3-85093-180-3.
- Helmut Kewitz, Helmut Coper, Diether Neubert, Eckard Oberdisse, Konrad Keller, Günter Schultz: Institut für Pharmakologie, Fachbereich Humanmedizin der Freien Universität Berlin. In: Athineos Philippu (Hrsg.): Geschichte und Wirken der pharmakologischen, klinisch-pharmakologischen und toxikologischen Institute im deutschsprachigen Raum, S. 47–58. Berenkamp-Verlag, Innsbruck 2004. ISBN 3-85093-180-3.
Einzelnachweise
- Manfred Lindinger, Verflochtene Signalwege: Zum Tod des Biochemikers Günter Schultz, In: Frankfurter Allgemeine Zeitung vom 20. August 2021
- G. Senft, M. Hoffmann, K. Munske, G. Schultz: Effects of hydration and dehydration on cyclic adenosine 3′,5′-monophosphate concentration in the rat kidney. In: Pflüger's Archiv für die gesamte Physiologie des Menschen und der Tiere. 298, 1968, S. 348–358. doi:10.1007/BF00363874.
- G. Schultz, G. Senft, W. Losert, R. Sitt: Biochemische Grundlagen der Diazoxid-Hyperglykämie. In: Naunyn-Schmiedebergs Archiv für experimentelle Pathologie und Pharmakologie. 253, 1966, S. 372–387. doi:10.1007/BF00245976.
- G. Senft, W. Losert, G. Schultz, R. Sitt, H. K. Barthelheimer: Ursachen der Störungen im Kohlenhydratstoffwechsel unter dem Einfluß sulfonamidierter Diuretica. In: Naunyn-Schmiedebergs Archiv für experimentelle Pathologie und Pharmakologie. 255, 1966, S. 369–382. doi:10.1007/BF00593171.
- E. Böhme, K. Munske, G. Schultz: Bildung von cyclischem Guanosin-3′,5′-monophosphat in verschiedenen Geweben der Ratte. In: Naunyn-Schmiedebergs Archiv für Pharmakologie. 264, 1969, S. 220–221. doi:10.1007/BF02431417.
- G. Schultz, E. Böhme, K. Munske: Guanyl cyclase. Determination of enzyme activity. In: Life Sciences. 8, Nr. 13, 1968, S. 1323–1332. doi:10.1016/0024-3205(69)90189-1. PMID 5363364.
- Doris Koesling, Eycke Böhme, Günter Schultz: Guanylyl cyclases, a growing family of signal-transducing enzymes. In: FASEB Journal. 5, Nr. 13, 1991, S. 2785–2791. PMID 1680765.
- Lincoln R. Potter: Guanylyl cyclase, structure and function. In: Cellular Signalling. 23, 2011, S. 1921–1926. doi:10.1016/j.cellsig.2011.09.001.
- Rupert Gerzer, Franz Hofmann, Günter Schultz: Purification of a soluble, sodium-nitroprusside-stimulated guanylate-cyclase from bovine lung. In: European Journal of Biochemistry. 116, 1981, S. 479–483. doi:10.1111/j.1432-1033.1981.tb05361.x. PMID 6114859.
- Rupert Gerzer, Eycke Böhme, Franz Hofmann, Günter Schultz: Soluble guanylate cyclase purified from bovine lung contains heme and copper. In: FEBS Letters. 132, 1981, S. 71–74. doi:10.1016/0014-5793(81)80429-2. PMID 6117479.
- Doris Koesling, Joachim Herz, Heinrich Gausepohl, Feraydoon Niroomand, Klaus-Dieter Hinsch, Alexander Mülsch, Eycke Böhme, Günter Schultz, Rainer Frank: The primary structure of the 70 kDa subunit of bovine soluble guanylate cyclase. In: FEBS Letters. 239, Nr. 1, 1988, S. 29–34. doi:10.1016/0014-5793(88)80539-8. PMID 2903071.
- Christian Harteneck, Barbara Wedei, Doris Koesling, Jürgen Malkewitz, Eycke Böhme, Günter Schultz: Molecular cloning and expression of a new a-subunit of soluble guanylyl cyclase. In: FEBS Letters. 292, 1991, S. 217–222. doi:10.1016/0014-5793(91)80871-Y. PMID 1683630.
- G. Schultz, J. G. Hardman, K. Schultz, J. W. Davis, E. W. Sutherland: A new enzymatic assay for guanosine 3′:5′-cyclic monophosphate and its application to the ductus deferens of the rat. In: Proceedings of the National Academy of Sciences. 70, 1973, S. 1721–1725. doi:10.1073/pnas.70.6.1721. PMID 4352651.
- G. Schultz, J. G. Hardman, K. Schultz, C. E. Baird, E. W. Sutherland: The importance of calcium ions for the regulation of guanosine 3':5'-cyclic monophosphate levels. In: Proceedings of the National Academy of Sciences. 70, Nr. 12, 1973, S. 3389–3393. doi:10.1073/pnas.70.12.3889. PMID 4359494.
- Klaus-Dieter Schultz, Karin Schultz, Günter Schultz: Sodium nitroprusside and other smooth muscle-relaxants increase cyclic GMP levels in rat ductus deferens. In: Nature. 265, 1977, S. 750–751. doi:10.1038/265750a0.
- Klaus-Dieter Schultz, Eycke Böhme, Volker A. W. Kreye, Günter Schultz: Relaxation of hormonally stimulated smooth muscular tissues by the 8-bromo derivative of cyclic GMP. In: Naunyn-Schmiedeberg's Archives of Pharmacology. 306, 1979, S. 1–9. doi:10.1007/BF00515586.
- K. H. Jakobs, W. Saur, G. Schultz: Reduction of adenylate cyclase activity in lysates of human platelets by the alpha-adrenergic component of epinephrine. In: Journal of Cyclic Nucleotide Research. 2, 1976, S. 381–392. PMID 14175.
- Karl H. Jakobs, Wilhelm Saur, Günter Schultz: Characterization of α- and β-adrenergic receptors linked to human platelet adenylate cyclase. In: Naunyn-Schmiedeberg's Archives of Pharmacology. 302, 1978, S. 285–291. doi:10.1007/BF00508297.
- Peter Lasch, Karl H. Jakobs: Agonistic and antagonistic effects of various α-adrenergic agonists in human platelets. In: Naunyn-Schmiedeberg's Archives of Pharmacology. 306, 1979, S. 119–125. doi:10.1007/BF00498981.
- Karl H. Jakobs, Wilhelm Saur, Günter Schultz: Inhibition of platelet adenylate cyclase by epinephrine requires GTP. In: FEBS Letters. 85, 1978, S. 167–170. doi:10.1016/0014-5793(78)81272-1. PMID 620788.
- Karl H. Jakobs, Klaus Aktories, Günter Schultz: GTP-dependent inhibition of cardiac adenylate cyclase by muscarinic cholinergic agonists. In: Naunyn-Schmiedeberg's Archives of Pharmacology. 310, 1979, S. 113–119. doi:10.1007/BF00500275.
- Lee E. Limbird: Activation and attenuation of adenylate cyclase. The role of GTP-binding proteins as macromolecular messengers in receptor--cyclase coupling. In: Biochemical Journal. 195, 1981, S. 1–13. PMID 6272740.
- Karl H. Jakobs, Klaus Aktories, Günter Schultz: A nucleotide regulatory site for somatostatin inhibition of adenylate cyclase in S49 lymphoma cells. In: Nature. 303, 1983, S. 177–178. doi:10.1038/303177a0. PMID 6140642.
- Klaus Aktories, Günter Schultz, Karl H. Jakobs: Islet-activating protein impairs α2-adrenoceptor-mediated inhibitory regulation of human platelet adenylate cyclase. In: Naunyn-Schmiedeberg's Archives of Pharmacology. 324, 1983, S. 196–200. doi:10.1007/BF00503894.
- Nina Wettschureck, Stefan Offermanns: Mammalian G proteins and their cell type specific functions. In: Physiological Reviews. 85, 2005, S. 1159–1204. doi:10.1152/physrev.00003.2005.
- Franz Hofmann: Wirkungen von Pharmaka auf den Organismus: allgemeine Pharmakodynamik. In: K. Aktories, U. Förstermann, F. Hofmann und K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie, S. 4–24. 11. Auflage, München, Elsevier GmbH 2013. ISBN 978-3-437-42523-3
- J. Hescheler, W. Rosenthal, W. Trautwein, G. Schultz: The GTP-bindung protein, Go, regulates neuronal calcium channels. In: Nature. 325, 1987, S. 445–447. doi:10.1038/325445a0.
- Kathleen Dunlap, George G. Holz, Stanley G. Rane: G proteins as regulators of ion channel function. In: Trends in Neurosciences. 10, 1987, S. 241–244. doi:10.1016/0166-2236(87)90166-4.
- J. Hescheler, W. Rosenthal, K. D. Hinsch, M. Wulfern, W. Trautwein, G. Schultz: Angiotensin II-induced stimulation of voltage-dependent Ca2+ currents in an adrenal cortical cell line. In: The EMBO journal. Band 7, Nummer 3, März 1988, S. 619–624, PMID 2456209, PMC 454365 (freier Volltext).
- W. Rosenthal, J. Hescheler, K. D. Hinsch, K. Spicher, W. Trautwein, G. Schultz: Cyclic AMP-independent, dual regulation of voltage-dependent Ca2+ currents by LHRH and somatostatin in a pituitary cell line. In: The EMBO journal. Band 7, Nummer 6, Juni 1988, S. 1627–1633, PMID 2458919, PMC 457146 (freier Volltext).
- David E. Clapham: Direct G protein activation of ion channels?. In: Annual Review of Neuroscience. 17, 1994, S. 441–464. doi:10.1146/annurev.ne.17.030194.002301.
- C. Kleuss, J. Hescheler, C. Ewel, W. Rosenthal, G. Schultz, B. Wittig: Assignment of G-protein subtypes to specific receptors inducing inhibition of calcium currents. In: Nature. 353, 1991, S. 43–48. doi:10.1038/353043a0.
- C. Kleuss, H. Scherübl, J. Hescheler, G. Schultz, B. Wittig: Different β-subunits determine G-protein interaction with transmembrane receptors. In: Nature. 358, Februar, S. 424–426. doi:10.1038/358424a0. PMID 1322501.
- Christiane Kleuss, Hans Scherübl, Jürgen Hescheler, Günter Schultz, Burghardt Wittig: Selectivity in signal transduction determined by γ subunits of heterotrimeric G proteins. In: Science. 259, 1992, S. 832–834. doi:10.1126/science.8094261. PMID 8094261.
- Maik Gollasch, Christiane Kleuss, Jürgen Hescheler, Burghardt Wittig, Günter Schultz: Gi2 and protein kinase C are required for thyrotropin-releasinghormone-induced stimulation of voltage-dependent Ca2+ channels in rat pituitary GH3 cells. In: Proceedings of the National Academy of Sciences. 90, 1993, S. 6265–6269. doi:10.1073/pnas.90.13.6265. PMID 8392194.
- Laugwitz habilitierte sich 2002 in München und wurde 2006 Leiter der I. Medizinischen Klinik des Klinikums rechts der Isar der Technischen Universität München.
- Anouk Allgeier, Stefan Offermanns, Jacqueline van Sande, Karsten Spicher, Günter Schultz, Jacques E. Dumont: The human thyrotropin receptor activates G-proteins Gs and Gq/11. In: Journal of Biological Chemistry. 269, 1994, S. 13733–13735. PMID 8188646.
- Karl-Ludwig Laugwitz, Anouk Allgeier, Stefan Offermanns, Karsten Spicher, Jacqueline van Sande, Jaques E. Dumond, Günter Schultz: The human thyrotropin receptor: A heptahelical receptor capable of stimulating members of all four G protein families. In: Proceedings of the National Academy of Sciences. 93, 1996, S. 116–120. doi:10.1073/pnas.93.1.116. PMID 8552586.
- Stefan Offermanns, Karl-Ludwig Laugwitz, Karsten Spicher, Günter Schultz: G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. In: Proceedings of the National Academy of Sciences. 91, 1994, S. 504–508. doi:10.1073/pnas.91.2.504. PMID 8290554.
- Birgit Klages, Ursula Brandt, Melvin I. Simon, Günter Schultz, Stefan Offermanns: Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. In: Journal of Cell Biology. 144, 1999, S. 745–754. doi:10.1083/jcb.144.4.745. PMID 10037795.
- Antje Gohla, Günter Schultz, Stefan Offermanns: Role for G12/G13 in agonist-induced vascular smooth muscle cell contraction. In: Circulation Research. 2000. doi:10.1161/01.RES.87.3.221. PMID 10926873.
- Thomas Gudermann, Frank Kalkbrenner, Günter Schultz: Diversity and Selectivity of receptor-G protein interaction. In: Annual Review of Pharmacology and Toxicology. 36, 1996, S. 429–459. doi:10.1146/annurev.pa.36.040196.002241.
- Craig Montell: The history of TRP channels, a commentary and reflection. In: Pflügers Archiv. 461, 2011, S. 400–506. doi:10.1007/s00424-010-0920-3. PMID 21287198.
- Christof Zitt, Andrea Zobel, Alexander G. Obukhov, Christian Harteneck, Frank Kalkbrenner, Andreas Lückhoff, Günter Schultz: Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. In: Neuron. 16, 1996, S. 1189–196. doi:10.1016/S0896-6273(00)80145-2. PMID 8663995.
- Stephen P. H. Alexander, Alistair Mathie, John A. Peters: Transient receptor potential (TRP) cation channels. In: British Journal of Pharmacology. 164, supplement 1, 2011, S. S166–S174. doi:10.1111/j.1476-5381.2011.01649_1.x.
- „Orai“ und „STIM“. Dazu: Lutz Birnbaumer: The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. In: Annual Review of Pharmacology and Toxicology. 49, 2009, S. 395–426. doi:10.1146/annurev.pharmtox.48.113006.094928.
- Thomas Hofmann, Alexander G. Obukhov, Michael Schaefer, Christian Harteneck, Thomas Gudermann, Günter Schultz: Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. In: Nature. 397, 1999, S. 259–263. doi:10.1038/16711. PMID 9930701.
- Rainer Strotmann, Christian Harteneck, Karin Nunnenmacher, Günter Schultz, Tim D. Plant: OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. In: Nature New Biology. 2, 2000, S. 695–702. doi:10.1038/35036318. PMID 11025659.
- Christian Harteneck, Tim D. Plant, Günter Schultz: From worm to man: three subfamilies of TRP channels. In: Trends in Neurosciences. 23, 2000, S. 159–166. doi:10.1016/S0166-2236(99)01532-5. PMID 10717675.
- David E. Clapham, David Julius, Craig Montell, Günter Schultz: International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. In: Pharmacological Reviews. 57, 2005, S. 427–450. doi:10.1124/pr.57.4.6.
- Mitgliedseintrag von Günter Schultz (mit Bild) bei der Deutschen Akademie der Naturforscher Leopoldina, abgerufen am 22. Juli 2016.