α-Oxidation
Die α-Oxidation ist ein alternativer biochemischer Abbaumechanismus für Fettsäuren, der zum Einsatz kommt, wenn die normalerweise angewandte β-Oxidation unmöglich ist. Dies ist der Fall, wenn eine Methylgruppe am Cβ deren dritten Schritt, die Oxidation zum Keton, verhindert. Der Name α-Oxidation beschreibt, dass das zur Carboxygruppe benachbarte (α-ständige) Kohlenstoffatom oxidiert wird.
| Übergeordnet |
| Fettsäureoxidation |
| Gene Ontology |
|---|
| QuickGO |
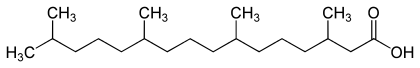
Bei Säugetieren wird Phytansäure als einzige Fettsäure auf diese Weise abgebaut. Diese entsteht durch Oxidation aus Phytol, einem Alkohol, der in veresterter Form in Chlorophyll zu finden ist. Im Gegensatz zu Menschen sind Wiederkäuer in der Lage, diesen Ester während der Darmpassage zu spalten; über deren Fleisch und Milch gelangen täglich etwa 100 mg in den menschlichen Körper. Aus energetischer Sicht spielt die α-Oxidation nur eine untergeordnete Rolle.[1]
Schrittweiser Ablauf
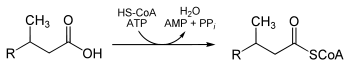
Aktivierung
Bevor der Abbau beginnen kann, muss die Säure an Coenzym A gebunden werden. Um die notwendige Energie für die Bildung des Thioesters zu liefern, wird Pyrophosphat von ATP abgespalten. Diese Reaktion katalysiert eine Phytanoyl-CoA-Ligase (EC 6.2.1.24):
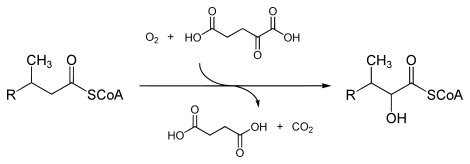
Abbau in den Peroxisomen
Das Enzym peroxisomale Phytanoyl-CoA-Dioxygenase[2] (EC 1.14.11.18) oxidiert das α-C-Atom der Phytansäure in Gegenwart von Ascorbinsäure und Eisen mit molekularem Sauerstoff zum Alkohol – das zweite Sauerstoffatom wird an 2-Oxoglutarat übertragen:
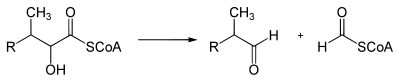
2-Hydroxyacyl-CoA-Lyase 1[3] katalysiert die Spaltung in Formyl-CoA und Pristanal; Cofaktoren sind Mg2+ und Thiaminpyrophosphat:
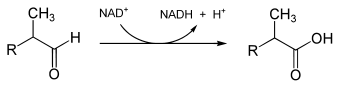
Vom Aldehyd, oder, genauer gesagt: von dessen Hydrat wird durch das Enzym Pristanal-Dehydrogenase[4] Wasserstoff abgespalten und an NAD⁺ übertragen:
Very-long-chain-Acyl-CoA-Synthetase[5] aktiviert die Säure erneut; die Energie stammt von ATP:
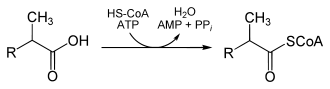
Diese Reaktion ist notwendig, um die entstandene Pristansäure aus den Peroxisomen zurück ins Cytoplasma zu befördern – sie kann in den Mitochondrien durch β-Oxidation vollständig zu 3 Acetyl-CoA, 3 Propionyl-CoA und Isobutyryl-CoA abgebaut werden.
Pathobiochemie
Sind entweder das Enzym peroxisomale Phytanoyl-CoA-Dioxygenase, das die Hydroxylierung am Cα katalysiert, oder Peroxin-7, ein Protein, das dieses Enzym in die Peroxisomen transportiert, defekt, so kann Phytansäure nicht abgebaut werden. Sie reichert sich folglich im Körper an und verursacht ein Refsum-Syndrom genanntes Krankheitsbild mit schweren neurologischen Problemen.[1] Dauerhafte Heilung gibt es nicht, durch eine phytansäurearme Diät können die Symptome der Krankheit wieder zurückgehen.
Einzelnachweise
- Thomas M. Devlin (Hrsg.): Textbook of Biochemistry with Clinical Correlations. Wiley & Sons; 6. Auflage 2006;ISBN 978-0-471-67808-3; S. 686
- UniProt O14832
- UniProt Q9UJ83
- noch nicht erfasst
- UniProt O14975
Siehe auch
Literatur
- Gerbert A. Jansen, Ronald J.A. Wanders: Alpha-Oxidation. In: Biochimica et Biophysica Acta - Molecular Cell Research. 1763, 2006, S. 1403, doi:10.1016/j.bbamcr.2006.07.012.
Weblinks
- Anthony S. Wierzbicki (2007): Peroxisomal disorders affecting phytanic acid alpha-oxidation: a review. In: Biochem Soc Trans. 35(Pt 5); 881–886; ISSN 0300-5127 PMID 17956237; PDF (freier Volltextzugriff, engl.)
- Ronald J. Wanders und Jasper C. Komen (2007): Peroxisomes, Refsum′s disease and the alpha- and omega-oxidation of phytanic acid. In: Biochem Soc Trans. 35(Pt 5); 865–869; ISSN 0300-5127 PMID 1795623; PDF (freier Volltextzugriff, engl.)