Pharmakologisches Chaperon
Pharmakologische Chaperone, auch Pharmacoperone genannt,[1][2] sind organische Verbindungen, die als reversible Inhibitoren spezifisch an ungefaltete Proteine binden und dadurch die Faltungsdynamik der Proteine in Richtung ihrer richtigen Konformation verschieben und diese stabilisieren.
Beschreibung
Falsch gefaltete Proteine sind die Ursache für eine Vielzahl von Erkrankungen. Zu den Proteinfehlfaltungserkrankungen (englisch protein misfolding diseases) gehören beispielsweise die Amyloidosen, Mukoviszidose, Amyotrophe Lateralsklerose, Phenylketonurie, Parkinson- und Alzheimer-Krankheit.[3] In den Zellen der betroffenen Patienten werden die fehlgefalteten Proteine im Rahmen der Proteinqualitätskontrolle im endoplasmatischen Retikulum ausgesondert und im Proteasom zerlegt. Das Fehlen dieser Proteine, die beispielsweise als Enzyme, Rezeptoren oder Botenstoffe wichtige Funktionen in den Zellen übernehmen, führt zu den entsprechenden Krankheitsbildern. Die Ursachen für die Proteinfehlfaltungen sind vielschichtig. Sie reichen von Genmutationen in Exons, die zu Veränderungen in der Aminosäuresequenz, also der Primärstruktur des Genproduktes führen, über Fehler bei der Transkription oder der Translation. Diese Fehler haben unmittelbare Einflüsse auf die Sekundär- und Tertiärstruktur, beziehungsweise auf die Proteinfaltungskinetik. Pharmakologische Chaperone können in den Vorgang der Proteinfaltung eingreifen und diesen positiv beeinflussen. Es handelt sich um kleine Moleküle (small molecules) die als chemische Faltungshilfen dienen. Dadurch wird die Faltungsdynamik eines Proteins in Richtung seiner richtigen Konformation verschoben und stabilisiert. Mit der korrekten Tertiärstruktur wird die Proteinqualitätskontrolle im endoplasmatischen Retikulum „bestanden“[4][5] und das Protein kann an seinem Bestimmungsort seine Funktion erfüllen.
Von den pharmakologischen Chaperonen sind die chemischen Chaperone zu unterscheiden. Chemische Chaperone haben auf die Proteinstruktur einen unspezifischen Effekt. Sie verhindern beispielsweise die Proteinaggregation ungefalteter Proteine, indem sie die Löslichkeit erhöhen. Im Gegensatz dazu binden die pharmakologischen Chaperone spezifisch an ungefaltete Proteine – im Idealfall nur an einen Proteintyp – und stabilisieren die Struktur des Proteins.[6][7]
Natürliche Chaperone sind zelleigene Proteine, die den Faltungsprozess neu synthetisierter Proteine unterstützen. Bei einer Überexpression können Ansammlungen teilweise fehlgefalteter Proteine entstehen, die als Proteinaggregate bezeichnet werden.
Beispiele
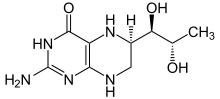
- Sapropterin ist zur Behandlung der Phenylketonurie (PKU) zugelassen. Ursache für die Erkrankung sind Mutationen im PAH-Gen, das für das Enzym Phenylalaninhydroxylase kodiert. Sapropterin stabilisiert als pharmakologisches Chaperon die Faltung einer Reihe von Mutationsvarianten der Phenylalaninhydroxylase.[3][8] Etwa 30 bis 50 % der PKU-Patienten sprechen auf Sapropterin an.[9] Sapropterin wurde im Dezember 2007 in den Vereinigten Staaten und im April 2009 in der Europäischen Union als erstes pharmakologisches Chaperon zur Behandlung zugelassen.[10]
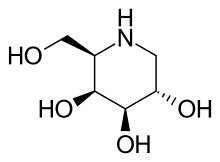
- Der Iminozucker 1-Deoxygalactonojirimycin (DGJ, internationaler Freiname Migalastat) ist ein pharmakologisches Chaperon. Es ist ein Analogon der terminalen Galactose von Gb3 und ein reversibler Inhibitor der α-Galactosidase A.[11] In einer Vielzahl von präklinischen Versuchen konnte gezeigt werden, dass Migalastat in der Lage ist, die Aktivität mutierter Varianten der α-Galactosidase A zu erhöhen.[12] Als kleines Molekül hat Migalastat eine sehr breite Bioverteilung im Organismus und kann beispielsweise das Zentralnervensystem erreichen und die Blut-Hirn-Schranke überwinden.[13] Darüber hinaus ist es oral verfügbar.[14] Migalastat befindet sich derzeit in der klinischen Phase III zur Erprobung der Wirksamkeit bei Patienten mit Morbus Fabry.[15]
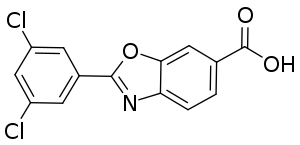
- Tafamidis (Handelsname Vyndaqel) ist ein pharmakologisches Chaperon zur Behandlung der Transthyretin-Amyloidose (ATTR). Das Protein Transthyretin liegt im Blut vor allem als Tetramer vor, das jedoch in Monomere zerfallen kann, die anfällig für Fehlfaltung sind. Bestimmte Mutationen des Transthyretin-Gens können diese Anfälligkeit verstärken. Tafamidis stabilisiert das Transthyretin-Tetramer durch Bindung an dessen Thyroxin-Bindungsstellen. Dadurch wird die Entstehung von Amyloiden verlangsamt, deren Ablagerung im Gewebe für die krankheitsbedingten Folgeschäden der ATTR verantwortlich ist.[16] Tafamidis ist in der EU zugelassen zur Behandlung der ATTR mit Kardiomyopathie (ATTR-CM) oder mit Polyneuropathie (ATTR-PN). Es kann sowohl bei genetisch bedingten Formen der ATTR wie der familiären Amyloidpolyneuropathie Typ I als auch bei Wildtyp-ATTR eingesetzt werden.[17][18]
Pharmakologische Chaperone könnten auch eine zukünftige Behandlungsmöglichkeit von Krankheiten durch Proteinaggregate wie der Alzheimer-Krankheit werden.[19][20]
Weiterführende Literatur
- P. M. Conn, J. A. Janovick: Pharmacoperone identification for therapeutic rescue of misfolded mutant proteins. In: Frontiers in endocrinology. Band 2, Nummer 6, März 2011, S. , ISSN 1664-2392. doi:10.3389/fendo.2011.00006. PMID 21633718. PMC 3103854 (freier Volltext).
- J. M. Benito, J. M. García Fernández, C. Ortiz Mellet: Pharmacological chaperone therapy for Gaucher disease: a patent review. In: Expert Opinion on Therapeutic Patents. Band 21, Nummer 6, Juni 2011, S. 885–903, ISSN 1744-7674. doi:10.1517/13543776.2011.569162. PMID 21457079 (Review).
- J. A. Janovick, B. S. Park, P. M. Conn: Therapeutic rescue of misfolded mutants: validation of primary high throughput screens for identification of pharmacoperone drugs. In: PLOS ONE. Band 6, Nummer 7, 2011, S. e22784, ISSN 1932-6203. doi:10.1371/journal.pone.0022784. PMID 21818389. PMC 3144936 (freier Volltext) (Open Access).
- R. S. Rajan, K. Tsumoto u. a.: Chemical and pharmacological chaperones: application for recombinant protein production and protein folding diseases. In: Current Medicinal Chemistry. Band 18, Nummer 1, 2011, S. 1–15, ISSN 1875-533X. PMID 21110818 (Review).
- S. K. Kalia, L. V. Kalia, P. J. McLean: Molecular chaperones as rational drug targets for Parkinson's disease therapeutics. In: CNS & Neurological Disorders – Drug Targets. Band 9, Nummer 6, Dezember 2010, S. 741–753, ISSN 1996-3181. PMID 20942788 (Review).
- Y. O. Ali, B. M. Kitay, R. G. Zhai: Dealing with misfolded proteins: examining the neuroprotective role of molecular chaperones in neurodegeneration. In: Molecules. Band 15, Nummer 10, 2010, S. 6859–6887, doi:10.3390/molecules15106859. PMID 20938400. PMC 3133442 (freier Volltext) (Review).
- J. A. Janovick, A. Patny u. a.: Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. In: Molecular endocrinology. Band 23, Nummer 2, Februar 2009, S. 157–168, ISSN 0888-8809. doi:10.1210/me.2008-0384. PMID 19095769. PMC 2646616 (freier Volltext).
- D. Williams, L. A. Devi: Escorts take the lead molecular chaperones as therapeutic targets. In: Progress in molecular biology and translational science. Band 91, 2010, S. 121–149, ISSN 1877-1173. doi:10.1016/S1877-1173(10)91005-3. PMID 20691961. PMC 3125676 (freier Volltext) (Review).
- M. A. Babizhayev: Designation of imidazole-containing dipeptides as pharmacological chaperones. In: Human & Experimental Toxicology. Band 30, Nummer 7, Juli 2011, S. 736–761, ISSN 1477-0903. doi:10.1177/0960327110377526. PMID 20656726 (Review).
- D. P. Germain, J. Q. Fan: Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: in vitro and preclinical studies. In: International Journal of Clinical Pharmacology and Therapeutics. Band 47 Suppl 1, 2009, S. S111–S117, ISSN 0946-1965. PMID 20040321 (Review).
- G. Parenti: Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. In: EMBO molecular medicine. Band 1, Nummer 5, August 2009, S. 268–279, ISSN 1757-4684. doi:10.1002/emmm.200900036. PMID 20049730 (Review).
- T. K. Chaudhuri, S. Paul: Protein-misfolding diseases and chaperone-based therapeutic approaches. In: The FEBS Journal. Band 273, Nummer 7, April 2006, S. 1331–1349, ISSN 1742-464X. doi:10.1111/j.1742-4658.2006.05181.x. PMID 16689923 (Review).
Einzelnachweise
- G. Sposny: Pharmacoperone. In: Laborjournal. Ausgabe 1, 2003.
- P. M. Conn, A. Ulloa-Aguirre u. a.: G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. In: Pharmacological reviews. Band 59, Nummer 3, September 2007, S. 225–250, ISSN 0031-6997. doi:10.1124/pr.59.3.2. PMID 17878512. (Review).
- D. Manstein: Forschungsbericht 2009. (PDF; 110 kB) Institut für Biophysikalische Chemie, S. 33.
- S. Ishii, H. H. Chang u. a.: Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. In: Journal of pharmacology and experimental therapeutics. Band 328, Nummer 3, März 2009, S. 723–731, ISSN 1521-0103. doi:10.1124/jpet.108.149054. PMID 19106170.
- R. Hamanaka, T. Shinohara u. a.: Rescue of mutant alpha-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. In: Biochimica et biophysica acta. Band 1782, Nummer 6, Juni 2008, S. 408–413, ISSN 0006-3002. doi:10.1016/j.bbadis.2008.03.001. PMID 18381081.
- S. Vogelbein: Das Qualitätskontrollsystem in post-ER Kompartimenten eukaryotischer Zellen am Beispiel des Vasopressin-V2-Rezeptors. Dissertation, FU Berlin 2009, S. 21.
- T. Arakawa, Y. Kita u. a.: Aggregation suppression of proteins by arginine during thermal unfolding. In: Protein and peptide letters. Band 13, Nummer 9, 2006, S. 921–927, ISSN 0929-8665. PMID 17100648.
- S. W. Gersting, M. Staudigl u. a.: Activation of phenylalanine hydroxylase induces positive cooperativity toward the natural cofactor. In: The Journal of biological chemistry. Band 285, Nummer 40, Oktober 2010, S. 30686–30697, ISSN 1083-351X. doi:10.1074/jbc.M110.124016. PMID 20667834. PMC 2945563 (freier Volltext).
- B. Wick-Urban: Trotz Ketonurie normal essen. In: Pharmazeutische Zeitung Online 15, 2007.
- LMU München: Molekulare Pädiatrie – Genetische Erkrankungen mit Proteinfaltung. Abgerufen am 14. Oktober 2011.
- N. Asano, S. Ishii u. a.: In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. In: European Journal of Biochemistry. Band 267, Nummer 13, Juli 2000, S. 4179–4186, ISSN 0014-2956. PMID 10866822.
- S. Biastoff, B. Dräger: Calystegines. In: G. A. Cordell (Hrsg.): The Alkaloids: Chemistry and Biology. S. 91. eingeschränkte Vorschau in der Google-Buchsuche
- G. Andreotti, M. R. Guarracino u. a.: Prediction of the responsiveness to pharmacological chaperones: lysosomal human alpha-galactosidase, a case of study. In: Orphanet Journal of Rare Diseases. Band 5, 2010, S. 36, ISSN 1750-1172. doi:10.1186/1750-1172-5-36. PMID 21138548. PMC 3016270 (freier Volltext). (Open Access).
- R. Khanna, R. Soska u. a.: The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. In: Molecular therapy. Band 18, Nummer 1, Januar 2010, S. 23–33, ISSN 1525-0024. doi:10.1038/mt.2009.220. PMID 19773742. PMC 2839206 (freier Volltext).
- Klinische Studie (Phase III): Study of the Effects of Oral AT1001 (Migalastat Hydrochloride) in Patients With Fabry Disease bei Clinicaltrials.gov der NIH
- Teresa Coelho, Giampaolo Merlini, Christine E Bulawa, James A Fleming, Daniel P Judge, Jeffery W Kelly, Mathew S Maurer, Violaine Planté-Bordeneuve, Richard Labaudinière, Rajiv Mundayat, Steve Riley, Ilise Lombardo, Pedro Huertas: Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. In: Neurol Ther. Band 5, Nr. 1, 2016, S. 1–25, doi:10.1007/s40120-016-0040-x, PMID 26894299, PMC 4919130 (freier Volltext).
- Fachinformation Tafamidis-Meglumin 20 mg Weichkapseln, Stand Oktober 2020. Abgerufen am 2. Juni 2021.
- Fachinformation Tafamidis 61 mg Weichkapseln, Stand Oktober 2020. Abgerufen am 2. Juni 2021.
- S. K. Dash: Future targeted disease modifying drugs for Alzheimer's disease. In: Recent patents on CNS drug discovery. Band 6, Nummer 1, Januar 2011, S. 65–76, ISSN 1574-8898. PMID 21073430. (Review).
- ADDF awards $210,300 to Amicus Therapeutics to evaluate PCs for treating Alzheimer's disease. In: news-medical.net Vom 7. Mai 2010