1-Deoxygalactonojirimycin
1-Deoxygalactonojirimycin (DGJ), internationaler Freiname Migalastat (Handelsname: Galafold, Hersteller: Amicus Therapeutics), ist ein reversibler kompetitiver Hemmer der Enzymklasse der Galactosidasen. Migalastat ist der erste oral wirksame Arzneistoff zur Behandlung der lysosomalen Speicherkrankheit Morbus Fabry. Die Substanz wirkt als pharmakologisches Chaperon, das heißt, durch Bindung an das krankhaft fehlerhaft gefaltete Enzym α-Galactosidase A wird der Prozess der Proteinfaltung in Richtung der richtigen Konformation verschoben. Infolgedessen kann das „richtig gefaltete“ Enzym seine Funktion ausüben.
| Strukturformel | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
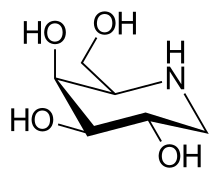 | |||||||||||||
| Allgemeines | |||||||||||||
| Freiname | Migalastat | ||||||||||||
| Andere Namen |
| ||||||||||||
| Summenformel | C6H13NO4 | ||||||||||||
| Kurzbeschreibung |
weißer kristalliner Feststoff[1] | ||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||
| |||||||||||||
| Eigenschaften | |||||||||||||
| Molare Masse | 163,17 g·mol−1 (freie Base) | ||||||||||||
| Aggregatzustand |
fest | ||||||||||||
| Löslichkeit |
als Hydrochlorid löslich in Wasser (≥ 1 mg/ml)[1] | ||||||||||||
| Sicherheitshinweise | |||||||||||||
| |||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||||||||
Beschreibung
Chemisch gesehen ist der Iminozucker 1-Deoxygalactonojirimycin ein Analogon der Galactose. Die Ähnlichkeit zu der endständigen Galactose-Gruppe des Moleküls Globotriaosylceramid (Gb3) ermöglicht die pharmakologische Wirkung in der Behandlung des Morbus Fabry.
Natürliches Vorkommen
Bisher wurde 1-Deoxygalactonojirimycin nicht in der Natur gefunden. Ein Derivat von DGJ, β-1-C-Butyl-DGJ, wurde im Jahr 2000 aus der Gattung Adenophora – einer Glockenblume – isoliert.[2] 1988 wurde 1-Deoxygalactonojirimycin erstmals aus der Fermentationsbrühe von Streptomyces lydicus PA-5726 gewonnen.[3][4]
Wirkungsmechanismus
Migalastat soll als pharmakologisches Chaperon die korrekte Faltung von Mutationsvarianten des Enzyms α-Galactosidase A ermöglichen. Morbus-Fabry-Patienten produzieren in ihren Zellen aufgrund einer Genmutation Varianten der α-Galactosidase A, die wegen ihrer falschen Faltung von der Proteinqualitätskontrolle im endoplasmatischen Retikulum (ER) ausgesondert und dem Proteasom zum Abbau zugeführt werden. In vielen Fällen könnten diese mutierten Enzymvarianten nicht nur die Proteinqualitätskontrolle bestehen, sondern auch ihrer Funktion als Enzym im Lysosom nachkommen, wenn sie korrekt gefaltet wären. Migalastat soll in diesen Prozess eingreifen, indem es als Faltungs-Templat der α-Galactosidase A dient. Dadurch soll die Faltungsdynamik des Proteins in Richtung der richtigen Konformation verschoben und stabilisiert werden. Mit der korrekten Tertiärstruktur wird die Qualitätskontrolle im ER „bestanden“.[5][6] Der stabile Chaperon-α-Galactosidase-A-Komplex wird durch Vesikel des endoplasmatischen Retikulums in den Golgi-Apparat und dann in das Lysosom transferiert. Dort soll Migalastat durch das natürliche Substrat (Gb3) ersetzt werden.[7] Die Dissoziation des Komplexes soll dabei durch die hohe Konzentration an Gb3 und den niedrigen pH-Wert im Lysosom begünstigt werden.[8]
Die EC50-Werte für α-Galactosidase A sind stark abhängig von der vorliegenden Mutationsvariante. Bei der In-vitro-Untersuchung von Lymphozyten mit 49 unterschiedlichen Missense-Mutationen variierten die EC50-Werte zwischen 820 nmol/L bis über >1 mmol/L.[9]
Klinische Anwendung
Anwendungsgebiet
Unter dem Handelsnamen Galafold wurde Migalastat im Mai 2016 von der EU-Kommission zugelassen zur oralen langfristigen Behandlung von Patienten ab 16 Jahren mit gesicherter Morbus-Fabry-Diagnose (α-Galaktosidase-A-Mangel), die eine auf die Behandlung ansprechende Mutation aufweisen.[10] Die unter den insgesamt mehr als 800 bekannten Mutationen im GLA-Gen für die Wirkung von Migalastat empfänglichen Mutationen wurden in einem proprietären in-vitro-Test ermittelt. Die aktuelle Zulassung umfasst alle 269 GLA-Mutationen, die auf Grundlage des Tests identifiziert und als ansprechend ermittelt wurden. Sie sind bei ungefähr 35 bis 50 Prozent Patienten mit Morbus Fabry nachzuweisen. Für nicht ansprechende Mutationen wird die Anwendung nicht empfohlen, ebenso ist Migalastat nicht vorgesehen für die gleichzeitige Behandlung zu einer Enzymersatztherapie.
Die amerikanische Zulassungsbehörde Food and Drug Administration (FDA) hat im August 2018 die Zulassung für Migalastat erteilt. Die Zulassung erfolgte im Rahmen eines beschleunigten Verfahrens (Accelerated Approval).[11]
Studien
Die Bewertung des Ausschusses für Humanarzneimittel, der im April 2014 die Zulassung empfahl[12], basiert auf klinischen Daten aus zwei Phase-III-Pivot-Studien an 127 Patienten sowie auf klinischen Ergebnissen aus einer unverblindeten Verlängerungsstudie. Die Morbus-Fabry-Patienten hatten eine GLA-Mutation, die auf Migalastat anspricht. Es wurden sowohl Patienten untersucht, die zuvor eine Enzymersatztherapie mit Agalsidase alfa/beta (Standardtherapie) erhalten hatten und auf Migalastat umgestellt wurden (ATTRACT-Studie), als auch Patienten, die zuvor noch keine Enzymersatztherapie erhalten hatten bzw. diese mehr als sechs Monate zurücklag (FACETS-Studie). Maßgeblich für die Beurteilung der Wirksamkeit war die Veränderung der Nierenfunktion der Patienten nach einer 18-monatigen Behandlung. Galafold erwies sich hinsichtlich der Stabilisierung der Nierenfunktion der Patienten als genauso wirksam wie die Enzymersatztherapie. Ferner verbesserte Galafold die Herzfunktion (Senkung des linksventrikulären Massenindex).
Nebenwirkungen und Anwendungsbeschränkungen
Als häufigste Nebenwirkung traten Kopfschmerzen auf, die bei etwa 10 % der Patienten, die Galafold erhalten hatten, beobachtet wurden.
Anwendungsbeschränkungen können sich ergeben, wenn eine Schwangerschaft besteht oder geplant ist, da Daten zur Wirkung von Migalastat auf das Ungeborene nicht ausreichend vorliegen. Ob Migalastat in die Muttermilch übertritt, ist nicht bekannt.
Synthese
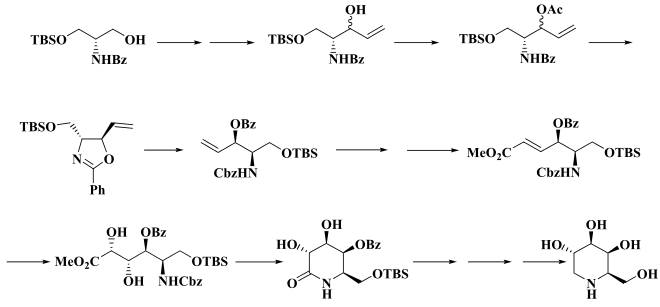
Die Totalsynthese von 1-Deoxygalactonojirimycin ist, für solch ein kleines Molekül, relativ aufwändig. Ein Syntheseweg geht D-N-Benzoylserinol, einem Derivat des Serinols (2-Amino-1,3-propandiol), aus und führt über mehrere Stufen zu einem trans-Oxazolin (Bildmitte, links), aus dem in etwa 25%iger Ausbeute das 1-Deoxygalactonojirimycin nach acht weiteren Stufen erhalten werden kann.[13] In der Literatur sind weitere Synthesewege beschrieben.[14] Eine elegante Synthese geht über insgesamt acht Stufen und hat eine Gesamtausbeute von 35 bzw. 43 %[15] bei über 99 % Enantiomerenreinheit.[16]
Handelsnamen
- Galafold (EU)
Weiterführende Literatur
- P. Compain, O. R. Martin (Hrsg.): Iminosugars: from synthesis to therapeutic applications. Verlag John Wiley and Sons, 2007, ISBN 0-470-03391-6 eingeschränkte Vorschau in der Google-Buchsuche
- K. Suzuki, T. Nakahara, O. Kanie: 3,4-Dihydroxypyrrolidine as glycosidase inhibitor. In: Current Topics in Medicinal Chemistry. Band 9, Nummer 1, 2009, S. 34–57, PMID 19199995. (Review).
- R. Khanna, R. Soska u. a.: The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. In: Molecular therapy : the journal of the American Society of Gene Therapy. Band 18, Nummer 1, Januar 2010, S. 23–33, doi:10.1038/mt.2009.220. PMID 19773742. PMC 2839206 (freier Volltext).
- E. R. Benjamin, J. J. Flanagan u. a.: The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. In: Journal of inherited metabolic disease. Band 32, Nummer 3, Juni 2009, S. 424–440, doi:10.1007/s10545-009-1077-0. PMID 19387866.
- S. Reißmann: Synthese und Glycosidase-Inhibitoreigenschaften von Calysteginen. Dissertation, Universität Halle-Wittenberg, 2006
Weblinks
- Öffentlicher Beurteilungsbericht (EPAR) der europäischen Arzneimittelagentur (EMA) zu: Migalastat
Einzelnachweise
- Datenblatt Deoxygalactonojirimycin hydrochloride bei Sigma-Aldrich, abgerufen am 4. Februar 2018 (PDF).
- K. Ikeda, M. Takahashi u. a.: Homonojirimycin analogues and their glucosides from Lobelia sessilifolia and Adenophora spp. (Campanulaceae). In: Carbohydrate Research. Band 323, Nummer 1–4, Januar 2000, S. 73–80, PMID 10782288.
- Y. Miyake, M. Ebata: The structures of a β-galactosidase inhibitor, Galactostatin, and its derivatives. In: Agric Biol Chem. Band 52, 1988, S. 661–666.
- N. Asano: Naturally occuring iminosugars and related alkaloids: structure, activity and applications. In: P. Compain, O. R. Martin (Hrsg.): Iminosugars: from synthesis to therapeutic applications. Verlag John Wiley and Sons, 2007, ISBN 0-470-03391-6, S. 17. eingeschränkte Vorschau in der Google-Buchsuche
- S. Ishii, H. H. Chang u. a.: Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. In: Journal of pharmacology and experimental therapeutics. Band 328, Nummer 3, März 2009, S. 723–731, doi:10.1124/jpet.108.149054. PMID 19106170.
- R. Hamanaka, T. Shinohara u. a.: Rescue of mutant alpha-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. In: Biochimica et biophysica acta. Band 1782, Nummer 6, Juni 2008, S. 408–413, doi:10.1016/j.bbadis.2008.03.001. PMID 18381081.
- S. Biastoff, B. Dräger: Calystegines. In: G. A. Cordell (Hrsg.): The Alkaloids: Chemistry and Biology. S. 91. eingeschränkte Vorschau in der Google-Buchsuche
- G. H. Yam, N. Bosshard u. a.: Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. In: American Journal of Physiology-Cell Physiology. 2006;290:S. C1076–C1082, doi:10.1152/ajpcell.00426.2005. PMID 16531566.
- E. R. Benjamin, J. J. Flanagan u. a.: The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. In: Journal of inherited metabolic disease. Band 32, Nummer 3, Juni 2009, S. 424–440, doi:10.1007/s10545-009-1077-0. PMID 19387866.
- Öffentlicher Beurteilungsbericht (EPAR) der europäischen Arzneimittelagentur (EMA) zu: 1-Deoxygalactonojirimycin
- FDA approves new treatment for a rare genetic disorder, Fabry disease, PM FDA vom 10. August 2018, abgerufen am 14. September 2018
- Galafold. Europäische Arzneimittel-Agentur. 1. April 2016.
- S. J. Pyun, K. Y. Lee u. a.: Synthesis Of (+)-1-Deoxygalactonojirimycin. In: Heterocycles. Band 62, 2004, S. 333–341.
- A. B. Hughes, A. J. Rudge: Deoxynojirimycin: synthesis and biological activity. In: Natural Product Reports. Band 11, Nummer 2, April 1994, S. 135–162, PMID 15209127. (Review).
- O. K. Karjalainen, A. M. P. Koskinen: Rapid and practical synthesis of (−)-1-deoxyaltronojirimycin. In: Org Biomol Chem. Band 9, 2011, S. 1231–1236. doi:10.1039/C0OB00747A
- O. K. Karjalainen, M. Passiniemi, A. M. Koskinen: Short and straightforward synthesis of (–)-1-deoxygalactonojirimycin. In: Organic letters. Band 12, Nummer 6, März 2010, S. 1145–1147, doi:10.1021/ol100037c. PMID 20170191.