Cockayne-Syndrom
Das Cockayne-Syndrom (CS; auch Neill-Dingwall-Syndrom) ist eine seltene, progrediente, autosomal-rezessiv vererbte Erkrankung. Sie ist hauptsächlich durch einen verhältnismäßig kleinen Kopf (Mikrozephalie), geringe Gewichtszunahme und ein beeinträchtigtes Wachstum charakterisiert.[1][2] Andere häufige Symptome sind eine verzögerte, mit dem Fortschreiten der Erkrankung rückläufige Entwicklung, Hör- und Sehminderung (fortschreitendem bilaterale Hörminderung bis zu Gehörlosigkeit, Katarakt, retinale Dystrophie, Photophobie), Muskelschwäche, Kontrakturen der Gelenke und Lichtempfindlichkeit der Haut (Photosensibilität). Erste Symptome sind in selten Fällen bereits pränatal oder kurz nach der Geburt vorhanden, bei den meisten betroffenen Individuen treten sie jedoch erst nach dem ersten Lebensjahr oder später in Erscheinung. Dies erschwert in den meisten Fällen eine frühe Diagnosestellung. Allgemein besteht eine hohe klinische Variabilität zwischen den einzelnen Betroffenen.[3][4][5]
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.1 | Angeborene Fehlbildungssyndrome, die vorwiegend mit Kleinwuchs einhergehen, inkl. Cockayne-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Namensherkunft
Das Cockayne-Syndrom ist nach dem englischen Arzt Edward Alfred Cockayne (1880–1956) benannt, welcher 1936 ein Geschwisterpaar beschrieb, das die für CS typischen Merkmale aufwies.[6] Zehn Jahre später wurde ein zweiter Artikel von Cockayne über die Geschwister veröffentlicht, in welchem er deren Entwicklung dokumentierte, sowie ausführlichere Informationen zu Familiengeschichte und Geburtsgewicht gab.[7] Im Jahr 1950 berichteten Catherine A. Neill und Mary M. Dingwall über zwei Brüder, welche vergleichbare Symptome wie die von Cockayne beschriebenen Geschwistern aufwiesen. In ihrem ausführlichen Artikel verglichen sie die Erkrankung aufgrund der Entdeckung von Kalkablagerungen im Gehirn der Betroffenen und anderer Anzeichen vorzeitiger Alterung mit dem Hutchinson-Gilford-Syndrom (Progerie) und dem Werner-Syndrom.[8]
Ursache
Das Cockayne-Syndrom zählt zusammen mit Xeroderma pigmentosum (XP) und Trichothiodystrophie (TTD) zu den Nukleotidexzisionsreparatur-Erkrankungen (DNA-Reparatur). Betroffene haben in der Regel eine Mutation im CSA-Gen (ERCC8-Gen auf Chromosom 5, Genlocus q12.1) oder CSB-Gen (ERCC6-Gen auf Chromosom 10, Genlocus q11). In sehr seltenen Fällen liegt eine Kombination aus einer CS-Mutation und einer XP-Mutation (XPB/ERCC3, XPD/ERCC2, XPG/ERCC5) vor.[3] (für einen Überblick zu Beziehungen zwischen NER-Faktoren und assoziierten Erkrankungen siehe die Veröffentlichung von Ferri, Orioli, & Botta, 2020.[9]). Diese Gene enthalten Information für die Proteinbiosynthese, welche an der Reparatur geschädigter DNA beteiligt sind. Wenn die DNA von Zellen geschädigt und nicht durch entsprechende Proteine repariert wird, sind die Zellen in ihrer Funktion eingeschränkt und sterben ab. Dieser Prozess ist ein Teil der normalen Alterung des Menschen.
Dass ein Defekt in der transkriptions-gekoppelte Nukleotidexzisionsreparatur (tc-NER) allein die Ursache des klinischen Krankheitsbildes ist, wird von verschiedenen Forschenden in Frage gestellt, da dieser nicht alle Merkmale von CS erklären kann.[2] Es wird vermutet, dass auch andere CS-Proteine eine Rolle spielen.[10] und diskutiert, ob mitochondriale Dysfunktionen und Defekte der Basenexzisionsreparatur ursächlich an der Entstehung des klinischen Krankheitsbildes beteiligt sind.[11][12]
Merkmale und Symptome
Abgesehen von der Mikrozephalie, der beeinträchtigten Gewichtszunahme und beschränktem Wachstum, welche bei allen Betroffenen auftreten und mit dem Fortschreiten der Erkrankung ein Plateau erreichen, auf dem sich das Wachstum einpendelt bzw. stoppt, gibt es eine Vielzahl von weiteren Merkmalen, welche auftreten können, aber nicht zwingend auftreten müssen und deren Beginn und Ausprägung sich zwischen den einzelnen Betroffenen teils stark unterscheidet.
Aussehen
Kinder mit Cockayne-Syndrom haben in der Regel keine Gesichtsfehlbildungen, die zu einer frühen Diagnose beitragen könnten. Zu Beginn ihres Lebens erscheinen bei manchen Kindern die Augen klein und eingesunken (Enophthalmie). Dieses Merkmal entwickelt sich mit der Zeit bei allen Betroffenen und steht häufig in Verbindung mit einem fortschreitenden Verlust von subkutanen und orbitalem Fettgewebe (Lipatrophie).[2][3] Dadurch können auch Nase, Kinn und Ohren besonders prominent erscheinen. Andere Autoren beschreiben Kieferfehlstellungen (wie Retrognathie und Mikrognathie), einen hoch gebogenen, verschmälerten Gaumen und verschiedene weitere dentale Merkmale wie Zahnengstand, Hypodontie, Mikrodontie, Radikulomegalie (sehr lange Zahnwurzeln) und Zahnschmelzhypoplasie.[13] Weitere beschriebene Merkmale sind trockene Haut sowie trockenes, dünnes bzw. lichtes Haar.[1][2][3]
Wachstumsstörung und Mikrozephalie
Ein eingeschränktes Wachstum ist meist das erste Symptom der Erkrankung. Der Beginn der Wachstumsverzögerung tritt in seltenen Fällen bereits vor der Geburt auf, meist aber im Laufe der ersten Lebensjahre oder später (siehe Klassifizierung). Zeitpunkt des Auftretens und Schweregrad bzw. Abweichung zum erwarteten Wachstum verlaufen in der Regel parallel zum Auftreten und Schweregrad anderer Symptome. Nach einer anfänglich rasch abflachenden Wachstumskurve – im Vergleich zu gesunden Gleichaltrigen – pendelt sich das Körpergewicht auf einen bestimmten Wert ein[14]. Da das Größenwachstum sich ebenfalls verlangsamt, kann trotz einer großen Abweichung zu Gleichaltrigen der BMI für leichter Betroffene im normalen oder unteren normalen Bereich liegen; für schwerer Betroffene liegt er dagegen in der Regel unter dem normalen Bereich.[5] Im späten Stadium der Erkrankung kann es zu einem moderaten Gewichtsverlust kommen.[3]
Eine fortschreitende Mikrozephalie ist ein Merkmal, das ebenfalls alle Erkrankten betrifft. Bei der Geburt liegt der Kopfumfang häufig noch innerhalb des normalen Bereichs oder im unteren normalen Bereich der Wachstumskurve. Vereinzelt kann die Mikrozephalie aber auch bereits bei der Geburt vorhanden sein. Durch ein reduziertes Wachstum kann der Kopfumfang von stärker betroffenen Individuen nach einiger Zeit unterhalb die dritte Perzentile der Wachstumskurve für das entsprechende Alter fallen.[3] Bei Betroffenen, bei denen erste Symptome hingegen erst später auftreten, kann das Wachstum bis zum 10. Lebensjahr unauffällig sein[5]
Sensorisches System: Hören und Sehen
Ein bilateraler, sensorineuraler Gehörverlust ist häufig bei CS. Der Zeitpunkt des Auftretens variiert und neurologische und kognitive Beeinträchtigungen können eine Diagnose erschweren. Es scheint ein Zusammenhang der Schwere des Gehörverlusts mit dem gesamten Schweregrad der Erkrankung zu bestehen.[3]
Eine Trübung der Augenlinse (Katarakt) tritt bei etwa der Hälfte der Betroffenen auf. Diese kann angeboren sein oder erst später in Erscheinung treten und ist in den meisten Fällen beidseitig.[2][3]
Eine erhöhte Lichtempfindlichkeit der Augen (Photophobie) betrifft ebenfalls etwa die Hälfte der Betroffenen.[2][3] Von einem verringerten oder fehlenden Tränenfluss und verkleinerten Pupillen mit Widerstand der Erweiterung wurde ebenfalls berichtet.[3] Eine Veränderung der Pigmentierung der Netzhaut (Netzhautdystrophie) wurde häufig als herausragendes Merkmal beschrieben, oft einhergehend mit einem sogenannten „Salz-und-Pfeffer-Fundus“. Dies scheint jedoch erst im späten Stadium klinisch relevant zu werden und parallel zum gesamten Schweregrad der Erkrankung zu verlaufen.[2][3]
Knochen, Gelenke und Muskulatur
Progrediente Muskel- und Gelenkversteifungen (Kontrakturen) sind ein häufiges Merkmal von CS-Betroffenen. Teilweise bestehen die Probleme bereits nach der Geburt, sie können aber auch erst später auftreten. Die unteren Extremitäten sind in der Regel stärker betroffen als die oberen.[2][3]
Eine Muskelhypotonie des Rumpfes tritt ebenfalls häufig auf, bei neurologisch stärker Betroffenen oftmals in Kombination mit einer Muskelhypertonie der Arme und Beine. Die Schwäche des Rumpfes begünstigt eine progrediente Verformungen der Wirbelsäule wie Skoliose und Kyphose, welche ebenfalls häufig beschrieben werden.[2][3] Die teilweise und bei neurologisch stärker Betroffenen vermehrt auftretende erhöhte Spannung in den Beinen kann zu Problemen mit der Hüfte (Hüftdysplasie bis Hüftsubluxation) beitragen.[3]
Teilweise wurde von Osteoporose bei älteren Betroffenen berichtet, aber es ist unklar, ob es sich dabei um ein spezifisches Merkmal oder die Konsequenz einer eingeschränkten Mobilität handelt. Eine Häufung von Knochenbrüchen wurde nicht beobachtet.[3]
Dermatologische Merkmale
Eine pathologische Lichtempfindlichkeit der Haut (Photosensibilität) galt lange als Hauptmerkmal von CS, vor allem aufgrund des zugrundeliegenden DNA-Reparaturdefekts und der daraus resultierenden Sensibilität der Zellen auf UV-Licht. Klinisch leiden jedoch nur etwa ein Drittel bis drei Viertel der Betroffenen an Photosensibilität.[2][3] Diese haben bereits nach kurzer Sonnenexposition einen leichten bis schweren Sonnenbrand. Bei manchen Individuen wird das Auftreten von pigmentierte Hautflecken auf sonnenexponierten Hautarealen beschrieben. Menschen mit einer CSA- oder CSB-Mutation haben kein erhöhtes Risiko für das Auftreten von Hautkrebs.[2][3] Individuen mit einer zusätzlichen Mutation in einem XP-Gen, die ebenfalls dem Cockayne-Syndrom zugeordnet werden, leiden unter einer extremen Form der Photosensibilität (Xeroderma pigmentosum) und erhöhtem Risiko für Hautkrebs.[3]
Teilweise wurde von einer fehlenden oder verminderten Schweißabsonderung, trockener, juckender Haut, einer subtilen Dystrophie der Nägel, von Atrophie der Haut, Livedo reticularis und Ödemen an den Extremitäten berichtet.[3]
Neurologische Merkmale
Es gibt eine Vielzahl von neurologischen Symptomen, die je nach Beginn und Schwere der Erkrankung bei einzelnen Betroffenen unterschiedlich stark auftreten. Bei Betroffenen mit einem frühen Beginn erster Symptome werden Hypertonie der Gliedmaßen und Spasmen beobachtet. Durch eine zunehmende neurologische Dysfunktion nimmt die Geschwindigkeit der Sehnenreflexe ab. Symptome, die durch Veränderungen des Kleinhirns ausgelöst werden und bei fast allen Betroffenen auftreten, sind beispielsweise Störungen in der Bewegungskoordination (Ataxie) und Muskelzittern (Tremor). Ataxie und Tremor können bei anfänglich normaler Entwicklung erste Anzeichen für die Erkrankung sein. Krampfanfälle wurden gelegentlich beschrieben und kommen im Vergleich zur gesamten Bevölkerung etwas häufiger vor.[2][3] Wenn Sprache erworben wurde, können Sprachstörungen auftreten.[3]
Zu Beginn des Lebens sind Magnetresonanztomographie- und Computertomographie-Aufnahmen des Gehirns selbst bei früh und stark Betroffenen meist unauffällig. Später können Verkalkungsprozesse an verschiedenen Orten im Gehirn, Anomalien der weißen Substanz (Dysmyelinisierung und Hypomyelinisierung), Verlust der weißen Substanz (Demyelinisierung), vergrößerte zerebrale Ventrikel und Atrophie auftreten.[2][3]
Entwicklungsverzögerung
Die Entwicklung der Betroffenen ist unterschiedlich stark beeinträchtigt (siehe Klassifizierung). Die Schwere der Beeinträchtigung steht in der Regel in Zusammenhang mit dem gesamten Schweregrad der Erkrankung. Bei allen Betroffenen kommt es im späten Stadium der Erkrankung zu einem neurologischen Abbau, welcher mit einer Verringerung der kognitiven Fähigkeiten einhergeht.[3][5]
Leber, Niere und Blutdruck
Die Leberenzyme von Betroffenen sind häufig leicht erhöht (Transaminasenanstieg), jedoch ohne klinische Bedeutsamkeit. Gelegentlich wurde von einer vergrößerten Leber berichtet.[3]
Von Problemen mit der Niere wurde gelegentlich berichtet, aber diese sind nicht gut dokumentiert. Gelegentlich wurde von Bluthochdruck und Proteinurie berichtet, welche zu progressiven Nierenversagen führen können. Von einem akuten nephrotischen Syndrom und akutem Nierenversagen wurde vereinzelt in späten Stadien der Erkrankung berichtet. Dabei ist häufig unklar, ob es die Folge von chronischer Nierenproblemen ist oder durch andere pathologischen Prozesse verursacht wurde.[2][3]
Immunsystem
Betroffene erkranken im Allgemeinen im Vergleich mit der allgemeinen Bevölkerung nicht häufiger an Infekten.[2] Im fortgeschrittenen Stadium der Erkrankung kann das Immunsystem aber durch Bluthochdruck oder Diabetes mellitus beeinträchtigt sein.[3][5]
Persönlichkeit
In der Regel werden CS-Betroffene als sehr fröhlich, aufgeschlossen und kontaktfreudig beschrieben.[3][5]
Weitere Symptome
Häufig anhaltend kalte Hände und Füße, unabhängig vom Schweregrad; Verdauungsstörungen sowohl in Form von Obstipation als auch dünnen Stühlen; Schlafstörungen (insbesondere bei neurologisch stärker Betroffenen); schwaches Schreien nach der Geburt bei Individuen mit frühem Beginn erster Symptome; wiederkehrende Atemwegserkrankungen, häufig aufgrund von Problemen der Schluckfähigkeit und Aspiration; gelegentlich veränderter Zuckerstoffwechsel und Diabetes mellitus, beginnend im zweiten Lebensjahrzehnt; gelegentlich Atherosklerose und Durchblutungsstörungen.
Progerie?
Manche Symptome wie der Verlust von subkutanem Fett, progredienter Hörverlust, kognitiver Abbau, Nierenerkrankungen, Atherosklerose, chronischer Bluthochdruck und Diabetes erinnern an spezifische Zeichen des normalen Alterns und treten bei CS Betroffenen zu einem deutlich früheren Zeitpunkt auf, als bei der restlichen Bevölkerung. Aber alle anderen neurologischen Merkmale, ebenso wie die Degeneration der retinalen Pigmentierung oder spezifische Anomalien der Zähne, gehören nicht zum üblichen klinischen Spektrum des Alterns. Ob diese Symptome die Konsequenz der gleichen pathophysiologischen Mechanismen bei CS wie bei normalem Altern sind, ist nicht geklärt.[3]
Klassifizierung
In der Literatur wird aufgrund des variablen Beginns und der unterschiedlichen Schwere der Symptome häufig zwischen drei verschiedenen Typen des Cockayne-Syndroms unterschieden. Es hat sich jedoch als schwierig erwiesen, die einzelnen CS-Typen eindeutig und klar voneinander abzugrenzen, weswegen sich CS eher als kontinuierliches Spektrum beschreiben lässt. Für die Diagnose, die Prognose und für klinischen Studien scheint eine Klassifizierung dennoch sinnvoll, um Betroffene in weitgehend homogene Gruppen einteilen zu können.[3] Aber auch innerhalb dieser Gruppen wurden unterschiedliche Schweregrade beschrieben und einzelne Individuen lassen sich nicht immer eindeutig einem bestimmten Typ zuordnen. Für alle Typen gilt, dass nach einer anfänglichen Entwicklung und Zunahme von Fähigkeiten und Fertigkeiten, diese sich auf einem bestimmten Level einpendeln und anschließend ein Verlust von Kompetenzen und Fähigkeiten folgt.[5]
Typ I oder klassisches CS, auch moderates/gemäßigtes CS:
Nach der Geburt sind Gewicht, Kopfumfang und Größe in der Regel normal oder im unteren normalen Bereich. Während des ersten oder zweiten Lebensjahrs flacht die Wachstumskurve zunehmend ab und führt zu der für CS typischen kleinen Statur, Mikrozephalie und geringem Körpergewicht. Betroffene erreichen üblicherweise die frühen Meilensteine der Entwicklung wie sich drehen, sitzen und selbstständiges Essen. Die motorische und geistige Entwicklung ist jedoch üblicherweise verzögert. Einzelne weniger stark betroffene Individuen lernen frei zu laufen, während andere nicht oder nur wenige Schritte gehen können. Progrediente Beugekontrakturen, Skoliose, Kyphose, Ataxie und Tremor können die motorische Entwicklung behindern und die Fähigkeit zu gehen und zu stehen geht häufig mit Fortschreiten der Erkrankung wieder verloren. Auch die Sprachentwicklung beginnt in der Regel später als üblich. Stärker Betroffene erlernen häufig nur wenige Wörter, während weniger stark betroffene Individuen kurze Sätze zu bilden oder sich gut mit Zeichen verständigen können. Die Pubertät beginnt in der Regel zum üblichen Zeitpunkt.[5]
Typ II oder early-onset CS (früher Beginn), auch schwerwiegendes/schweres CS:
Wachstum und Gewichtszunahme sind im Vergleich zu den anderen Typen am stärksten beschränkt und erreichen ein frühes Plateau. In manchen Fällen wird ein zu geringes Wachstum (SGA oder IUGR) und/oder Mikrozephalie bereits in der Schwangerschaft beobachtet. Besondere Merkmale sind unter anderem früh auftretende und zunehmende Probleme bei der Nahrungsaufnahme, Schluckbeschwerden und eine eingeschränkte Entwicklung von motorischen und geistigen Fähigkeiten. Sehr stark Betroffene können sich nicht vom Rücken auf den Bauch drehen und erlernen weder sitzen, noch krabbeln. Schwächer betroffene Individuen erreichen diese ersten Meilensteine, verlieren die Fähigkeit frei zu sitzen in der Regel wieder. Das Ausbleiben des Lernens oder der Verlust dieser grundlegenden motorischen Fertigkeiten wird auch durch die Schwäche des Rumpfes, eine gesteigerte Spannung in den Armen und insbesondere den Beinen, fortschreitende Kontrakturen der Gelenke und Kyphose begünstigt. Die Sprachentwicklung ist in der Regel sehr beschränkt, manche weniger stark Betroffene erlernen einzelne Wörter oder Zeichen.[5]
Typ III oder mildes/atypisches CS:
Der Beginn erster Symptome, Krankheitsverlauf und Lebenserwartung variieren in dieser Gruppe am stärksten zwischen den einzelnen betroffenen Individuen. Die anfängliche Entwicklung ist in der Regel unauffällig und erste Symptome treten erst nach dem 3. Lebensjahr auf. Körpergröße und Gewicht können bis in das erste Lebensjahrzehnt hinein normal oder sogar über dem Durchschnitt liegen. Der Kopfumfang kann zwar etwas verringert, aber dennoch in der normalen statistischen Bandbreite wachsen, bleibt jedoch schließlich hinter dem Wert der durchschnittlichen Bevölkerung zurück. Das Wachstum des Kopfes stoppt üblicherweise zwischen dem 7 und 10. Lebensjahr. Dass dieser Typ als selten bzw. atypisch gilt, könnte daran liegen, dass Betroffene potentiell häufiger eine späte oder sogar keine CS Diagnose bekommen. Durch eine relativ normale motorische Entwicklung können Betroffene laufen und bis auf wenige Ausnahmen rennen und sportliche Aktivitäten ausüben. Die Entwicklung feinmotorischer Fertigkeiten ist jedoch häufig durch das frühe Auftreten eines Tremors eingeschränkt. Die Betroffenen lernen in der Regel in ganzen Sätzen sprechen sowie lesen und schreiben. Jedoch ist die geistige Entwicklung oftmals eingeschränkt und es wird von kognitiven Defiziten und verminderter Intelligenz berichtet. Die Pubertät tritt zum üblichen Zeitpunkt auf und es wurden vereinzelt erfolgreiche Schwangerschaften dokumentiert. Nach der anfänglich normal erscheinenden Entwicklung beginnt das Wachstum und die Gewichtszunahme zu stagnieren und es treten häufig ein Verlust des Appetits und zunehmende Schluckbeschwerden auf. Wenn die Erkrankung voranschreitet, kommt es zu einem Rückgang der motorischen und geistigen Fähigkeiten und es treten andere CS typische Symptome wie teilweiser oder vollständiger Hörverlust und Sehminderung in Erscheinung.[5] Auch von einer frühen Demenz, typischerweise nach dem 30. Lebensjahr, wurde berichtet.[3]
Diese klassischen Subtypen wurden von verschiedenen Autoren um zwei weitere Typen erweitert. So kann das COFS-Syndrom als besonders schwere Variante des CS-Syndroms angesehen werden, bei welcher sich die Symptome bereits vor der Geburt manifestieren. Daneben gibt es Individuen, bei welchen erste Symptome erst im Erwachsenenalter auftreten und es wurden Fälle beschrieben, bei welchen eine CSB-Mutation festgestellt wurde, aber abgesehen von einer ungewöhnlich starken Sensitivität für UV-Licht (UV-sensitives Syndrom) keine anderen für das CS-Syndrom typischen Symptome auftreten Diese beiden Varianten können als Extremfälle innerhalb eines kontinuierlichen CS-Spektrums angesehen werden.[3][5]
Es ist nicht geklärt, ob eine Beziehung zwischen Genotyp und Phänotyp besteht, also ob eine Mutation auf dem CSA- oder CSB-Gen die Entstehung eines bestimmten klinischen Krankheitsbildes bzw. Typs begünstigt. Bei stärker und früher Betroffenen scheinen jedoch etwas häufiger eine CSB Mutation und bei schwächer Betroffenen eine CSA-Mutation vorzuliegen.[3] Es könnten auch andere genetische und/oder Umweltfaktoren dabei eine Rolle spielen, welcher Schweregrad des CS Phänotyps sich bei einem einzelnen Individuen manifestiert.[1][3][15]
Es wurde angemerkt, dass für die Familien der Betroffenen eine Klassifizierung nicht unbedingt sinnvoll ist, da der Verlauf bei einzelnen Individuen sehr unterschiedlich sein kann (z. B. früher Beginn, jedoch eher langsames Voranschreiten der Symptome) und eine Zuteilung des erkrankten Kindes zu einem der beschriebenen Typen potentiell Unsicherheit hervorruft.[2]
Diagnose
Photosensibilität der Haut galt lange als das wichtigste diagnostische Kriterium. Vor der Entdeckung der für CS bedeutsamen Gene (CSA und CSB) Mitte der 1990er Jahre wurde auf molekularer Ebene ausschließlich die Testung der Sensitivität der Fibroblasten auf UV-C-Bestrahlung als Nachweis für eine CS-Erkrankung genutzt. Allerdings wurde die Rolle dieses Merkmals infrage gestellt und es wurde angemerkt, dass durch die alleinige Nutzung dieses Verfahrens als diagnostisches Kriterium einige Fälle in der Vergangenheit potentiell übersehen wurden. In einer Untersuchung zeigte sich, dass nur etwa ein Drittel der betroffenen Individuen unter einer ausgeprägten Photosensibilität litten und etwa 1/5 keine Zeichen für Photosensibilität zeigten.[2] Dies stimmt mit Berichten anderer Autoren überein, welche von einer Auftretenshäufigkeit von Photosensibilität bei CS-Betroffenen von etwa zwei Dritteln bis drei Vierteln berichten.[3]
Allgemein ist es oft schwierig, eine frühe Diagnose zu erhalten, da es sich um eine Erkrankung mit fortschreitendem Verlauf handelt und der Beginn sowie die Ausprägung der einzelnen Symptome zwischen betroffenen Individuen sehr unterschiedlich sein kann. Es wurden von verschiedenen Autoren Listen von Kriterien erstellt, die zur Diagnostik herangezogen werden können.
| Diagnostische Kriterien | Nance und Berry, 1992[1] | Laugel, 2013[3]* | Wilson, 2016[2]** |
|---|---|---|---|
| Hauptkriterien
müssen zwingend vorliegen |
- Entwicklungsverzögerung
- Wachstumsstörung/Minderwuchs |
- Entwicklungsverzögerung
- Progrediente Wachstumsstörung - Progrediente Mikrozephalie |
- Wachstumsstörung
- Mikrozephalie |
| Nebenkriterien
(mindestens 3 von 5 / mindestens 3 von 5 / |
- Lichtempfindlichkeit der Haut
- Netzhautdegeneration (Retinitis pigmentosa) und/oder Katarakt - Schallempfindungsschwerhörigkeit (sensorineurale Schwerhörigkeit) - Karies - Kachektischer Kleinwuchs |
- Lichtempfindlichkeit der Haut
- Netzhautdegeneration (Retinitis pigmentosa) und/oder Katarakt - Schallempfindungsschwerhörigkeit (sensorineurale Schwerhörigkeit) - Eingesunkene Augen (Enophthalmie) |
- Anhaltend kalte Hände und Füße
- Bilateraler Gehhörverlust - Lichtempfindlichkeit der Haut - (Intensions-) Tremor - Kontrakturen der Gelenke - Fortschreitender Verlust von subkutanem Fettgewebe - Katarakt - typische Gesichtsmerkmale |
| Ausschlusskriterien /
CS Diagnose in Frage stellende Kriterien |
- Fehlende Mikrozephalie | - Fehlbildung von Herz oder Nieren |
*Laugel, 2013: Zusätzlich können folgende Ergebnisse bildgebender Verfahren hilfreich für die Diagnosestellung sein: Hypomyelinisierung der weißen Substanz und Atrophie, Kleinhirnatrophie oder Hypoplasie und bilaterale Verkalkung des Putamen.
**Wilson, 2016: Entwicklungsverzögerung allein scheint ein eher schwaches Kriterium für die Diagnostik darzustellen, insbesondere für Betroffene mit einem späten Beginn erster Symptome. Bei Vorliegen der zwei Hauptkriterien und mindestens zwei der Nebenkriterien sollte wenn möglich direkt eine DNA-Analyse erfolgen, da für die Untersuchung der Erholung der RNA-Synthese der Fibroblasten nach UV-Bestrahlung eine Biopsie der Haut vorgenommen werden muss und das Ergebnis nicht immer eindeutig ist. Eine molekulargenetische Testung erfordert einen weniger invasiver Eingriff (Blut oder Speichelprobe) und liefert einen eindeutigen Befund.
Behandlung
Zum aktuellen Zeitpunkt (Stand: 2020) gibt es keine ursächliche Behandlung für die Erkrankung. Behandlungen zielen auf die Erhaltung einer möglichst hohen Lebensqualität für CS-Betroffenen ab und dienen der Vermeidung und Linderung schmerzhafter Symptome.
Im Fokus stehen dabei meist die reduzierte Gewichtszunahme, Hör- und Sehminderung, eine physiotherapeutische Behandlung und Versorgung mit Hilfsmitteln.
Reduzierte Gewichtszunahme
Das Wachstum und die Gewichtszunahme von Kindern mit CS fällt häufig bereits bevor eine Diagnose gestellt wurde unter die altersentsprechenden Perzentilen. Wenn eine Diagnose vorliegt, sollte bei der Entscheidung welche Maßnahmen ergriffen werden berücksichtigt werden, dass eine geringe Gewichtszunahme und ein eingeschränktes Wachstum ein grundlegender, nicht veränderbarer Teil der Erkrankung ist und die Entscheidung zu einer ergänzende Nahrungszufuhr sollte sich nach dem klinischen Status des Individuum richten.[2] Es wurden spezielle Wachstums- und Gewichtskurven für Kinder mit Cockayne-Syndrom veröffentlicht, welche für die Einschätzung ob und inwiefern eine Intervention notwendig ist, herangezogen werden können.[14]
Schluckschwierigkeiten sind häufig, besonders bei neurologisch stärker betroffenen Individuen und im fortgeschrittenen Stadium der Erkrankung kann Appetitlosigkeit auftreten. In manchen Fällen können daher Nahrungsergänzungsmittel wie normo- oder hochkalorische Trinknahrung hilfreich sein.[5] Besonders bei jungen Betroffenen kann es zu wiederkehrenden Erbrechen kommen, was eine wiederholte Gabe von kleinen Mengen erfordert.[3] Bei sehr schwer Betroffenen oder im fortgeschrittenen Stadium der Erkrankung kann eine transnasale Magensonde oder eine PEG-Sonde hilfreich für die Betroffenen sein.[2][3] Diese erleichtert ebenfalls die Gabe von Medikamenten. Ein bereits begonnener Abbau von subkutanem Fettgewebe kann durch eine Steigerung der Nahrungszufuhr oder Kalorienmenge nicht aufgehalten werden. In diesem Stadium der Erkrankung scheint der Magen Schwierigkeiten damit zu haben sich einer gesteigerte Nahrungsmenge anzupassen, was wiederum zu Erbrechen führen kann. Eine Steigerung der Nahrungsmenge sollte daher nur mäßig und gut überwacht stattfinden.[2] Bei kalorimetrischen Messungen in einer noch nicht publizierten Studie[16] (Stand:2020) wurde beobachtet, dass für viele Betroffene ein verringerter Kalorienbedarf besteht (bis zu 1/3 weniger als für die entsprechende Gewichts bzw. Altersklasse üblich) und daher die Hypothese überprüft, dass CS-Betroffene generell einen verminderten Stoffwechsel (Hypometabolismus) haben.
Verdauung
CS-Betroffene leiden häufig unter Verdauungsstörungen, welche sich sowohl in Form von Obstipation als auch dünnen Stühlen äußern kann. Ersteres kann medikamentös gut in den Griff bekommen werden. Bei wiederkehrenden Durchfällen ist auf einen Ausgleich des erhöhten Flüssigkeitsverlust und die Gefahr des Auftretens einer Windeldermatitis zu achten.
Hör- und Sehschwierigkeiten
Welche Behandlung bei Auftreten von Hör- und Sehschwierigkeiten sinnvoll ist, muss individuell entschieden werden. Häufig werden CS-Betroffene mit Hörgeräten versorgt, in manchen Fällen auch mit Cochlea-Implantaten. Katarakte können operativ entfernt werden. Je nach augenärztlichem Befund kann eine Brille verordnet werden. Generell wird empfohlen die Augen durch das Vermeiden direkter Sonne und das Tragen einer Sonnenbrille im Freien zu schützen.[2]
Physiotherapeutische und orthopädische Behandlung
Aufgrund der häufig auftretenden Probleme des Bewegungsapparats ist eine regelmäßige physiotherapeutische und orthopädische Begutachtung sinnvoll. Eine physiotherapeutische Behandlung sollte an die spezifischen Probleme des einzelnen Individuums angepasst stattfinden, um der Verschlechterung der Beweglichkeit, dem Muskelabbau und der Versteifung der Gelenke und Muskeln entgegenzuwirken. Der Einsatz von Hilfsmitteln wie Orthesen, Therapiestuhl, Reha-Buggy und Stehständer zur Unterstützung der Behandlung und zur Erleichterung des Alltags, sollte auf den Bedarf der Betroffenen abgestimmt werden.
Kontrakturen der Beine können auch zu Problemen mit der Hüfte führen, wodurch die Pflege von betroffenen Kindern erschwert werden kann (z. B. das Wickeln durch eine Abspreizhemmung der Beine). Durch eine geeignete physiotherapeutische Behandlung und den Einsatz von Hilfsmitteln kann diesem Problem frühzeitig entgegengewirkt werden. Teilweise führte der Einsatz von Botox-Injektionen in die Beine[5] oder Operationen zu einer Erleichterung dieses Problems. Die zu erwartenden Vorteile invasiver Maßnahmen sollte aber sehr gut abgewägt und nur wenn unbedingt erforderlich durchgeführt werden.[2][3]
Lichtempfindlichkeit der Haut
Auch für die eventuell vorhandene Photosensibilität der Haut gibt es keine ursächliche Behandlung. Bei besonders stark betroffenen Individuen sollten geeignete präventive und therapeutische Maßnahmen mit den behandelnden Ärzten/Ärztinnen besprochen werden. Für leichter betroffene Individuen sind in der Regel einfache Maßnahmen wie geeigneter Sonnenschutz durch entsprechende Kleidung, das Tragen eines Sonnenhuts und Sonnencreme ausreichend.[2]
Tremor
Durch frühe Auftreten eines Intensionstremor kann die feinmotorische Entwicklung, Aktivitäten wie Spielen oder Essen und so die gesamte Lebensqualität beeinträchtigt sein. Es wurde von guten Reaktionen auf Levodopa / Carbidopa berichtet.[2] Ob eine medikamentöse Behandlung in Frage kommt, muss individuell entschieden werden.
Dentale Probleme
Es wurde beobachtet, dass CS-Betroffene häufig unter Karies leiden. Dies könnte eventuell die Folge einer Zahnschmelzhypolasie, vermindertem Speichelfluss, gastroösophagealem Reflux, vermehrtem Erbrechen oder schlechter dentaler Hygiene aufgrund der neurologischen Beeinträchtigung sein.[3] Unabhängig von der Ursache ist es wichtig zu wissen, dass Karies unbehandelt zu Entzündungen mit starken Schmerzen führen kann und es sinnvoll den Zustand der Zähne regelmäßig begutachten zu lassen und gegebenenfalls frühzeitig Maßnahmen zur Vorbeugung oder Behandlung zu ergreifen.
Empfehlungen für die ärztliche Betreuung
In Deutschland wird die fortlaufende Betreuung von betroffenen Kindern optimaler Weise, zusätzlich zu einem niedergelassenen Kinderarzt/-ärztin, durch ein wohnortnahes sozialpädiatrisches Zentrum (SPZ) übernommen. Es wird empfohlen in regelmäßigen Abständen folgende Untersuchungen durchzuführen, um den Verlauf zu beobachten und gegebenenfalls rechtzeitig geeignete Maßnahmen für eine Behandlung ergreifen zu können:[2][17]
| System | Untersuchung | Häufigkeit |
|---|---|---|
| Ernährung | Begutachtung des Ernährungszustandes | Halbjährlich |
| Augen | Katarakte und Retinopathie | Halbjährlich bis zum 4. Lebensjahr, anschließend jährlich |
| Gehör | Dem Alter angepasste Hörtests | Jährlich |
| Klinische Untersuchungen von Herz-Kreislauf-System, Leber, Nieren, Stoffwechsel | - Blutdruck, weitere Untersuchungen wenn symptomatisch
- Leberenzyme - Nierenfunktion; Harnsäure & Proteinurie - Blutzuckerwerte (ab dem 10. Lebensjahr) |
Jährlich |
| Nervensystem | Neurologische/Neuropädiatrische Begutachtung | Halbjährlich |
| Bewegungsapparat | Orthopädische und physiotherapeutische Begutachtung | Halbjährlich/Jährlich |
Medikamente und stationäre Behandlung
Es wurde beobachtet, dass die Gabe von Metronidazol akutes Leberversagen hervorruft, welches tödlich verlaufen kann.[18] Auch verwandte Antibiotika sollten vorsichtshalber vermieden werden.[2]
In einzelnen Fällen wurde von einer ausgeprägten Reaktion auf Sedativa und Opioiden berichtet – diese reicht von Atemdepression bis zu emotionaler Abstumpfung über mehrere Tage nach der Gabe von Kodein. Für Opioide wird empfohlen, wenn nötig, mit einem Drittel der regulären Dosis zu beginnen.[2]
Andere Medikamente sollten stets nach Gewicht und nicht nach Alter dosiert werden.[2]
Die meisten CS-Betroffenen haben eine schlechte periphere Durchblutung und es kann schwierig sein einen Venenzugang zu legen; daher empfiehlt es sich, dass dies nur von besonders erfahrenen Personen durchgeführt wird.[2] Es wurde auch von Problemen bei der Intubation berichtet.[1] Wenn möglich sollten Erwachsene mit CS aufgrund der kleinen Statur und des geringen Gewichts (oftmals nur 10–17 kg[5][19]) in einer pädiatrischen Abteilung behandelt werden.[2]
Eine zu hohe intravenöse bzw. rasch ansteigende Flüssigkeitsgabe kann zu einer Überwässerung mit schwerwiegenden gesundheitlichen Folgen führen.[20] Es empfiehlt sich die intravenöse Flüssigkeitsgabe nicht nach Alters- bzw. Gewichtstabellen, sondern an der vorherigen Flüssigkeitsaufnahme zu orientieren und eine Steigerung der Flüssigkeitszufuhr nur sehr langsam und gut überwacht durchzuführen.
Zur Einschätzung des Ernährungszustandes wurden Wachstums- und Gewichtskurven für Kinder mit Cockayne-Syndrom veröffentlicht.[14]
Prognose
Durch progressiven Verlauf der Erkrankung und fehlende Behandlungsmöglichkeit der Ursache von CS ist die Lebenserwartung betroffener Individuen unterschiedlich stark eingeschränkt. Aufgrund der hohen Variabilität ist es schwierig eine pauschale Prognose für einzelne Individuen zu geben.
In einem systematischen Review von 1992 betrug das durchschnittliche Alter von 37 Betroffenen zum Zeitpunkt des Todes 12 Jahre; von diesen wurden drei Betroffene weniger als 2 Jahre und zwei Betroffene älter als 30 Jahre alt;[1] in einer Erhebung von 2016 betrug das durchschnittliche Alter von 28 Betroffenen zum Zeitpunkt des Todes 8,4 Jahre mit einer Bandbreite von 17 Monaten bis 30 Jahren.[2] In beiden Untersuchungen zeigte sich das Auftreten von Katarakten vor dem dritten Lebensjahr als wichtigster prognostischer Faktor für eine geringe Lebenserwartung. In einer anderen Untersuchung mit 45 Individuen zeigte sich jedoch kein solcher Zusammenhang; obwohl Katarakte häufiger bei schwer betroffenen Individuen auftraten, hatten zwei der ältesten Studienteilnehmenden (42 und 44 Jahre) bereits bei der Geburt Katarakte.[5]
Aufgeschlüsselt nach den drei verschiedenen Schweregraden von CS wurde die durchschnittliche Lebenserwartung für Betroffene des Typ I mit 16,1 Jahren (Bandbreite 11–22 Jahre), des Typ II mit 5 Jahren (Bandbreite 0,6–11 Jahre) und des Typ III mit 30,3 Jahren (Bandbreite 22–42 Jahre) berichtet.[5] In der Literatur gibt es jedoch auch Berichte von leicht betroffenen Individuen die 50 Jahre und älter wurden.[19]
Zu den vielfältigen Todesursachen gehören Pneumonie/Atemwegserkrankungen, Atemstillstand, Nieren- oder Leberversagen, Herzversagen, Komplikationen bei Krampfanfällen, Schlaganfall und weitere Komplikationen der verschiedenen Symptome Erkrankung.[3][5][21]
Häufigkeit
Die Auftretenshäufigkeit (Inzidenz) von CS in Westeuropa wird auf 2,7 pro eine Million Geburten geschätzt, könnte jedoch höher liegen.[22] Ein Grund für eine mögliche Unterschätzung der Auftretenshäufigkeit in der Vergangenheit könnte allgemein an der Weiterentwicklung diagnostischer Verfahren liegen[22] und darin, dass die Photosensibilität der Haut lange als wichtigstes diagnostisches Merkmal galt, wodurch einige Fälle ohne dieses Merkmal möglicherweise übersehen wurden.[2] Auch für Fälle mit einem späten Beginn erster Symptome kann es vorkommen, dass CS als die Ursache der Symptome lange unerkannt bleibt.[5]
In einer Untersuchung in Japan wurde die Prävalenz auf 1 zu 2,5 Millionen geschätzt.[21] Weibliche und männliche Individuen sind gleich häufig betroffen und CS ist weltweit verbreitet, wobei teilweise regionale Häufungen beobachtet wurden.[23][24]
Risiko für Familienangehörige
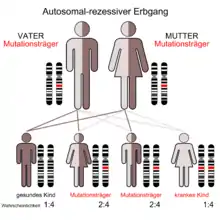
Die Eltern von betroffenen Kindern sich heterozygote Träger für eine pathogene Variante des CSA/ERCC8- oder CSB/ERCC6-Gens. Sie sind asymptomatisch und haben kein Risiko selbst die Erkrankung zu entwickeln. Die Geschwister von Betroffenen und die Geschwister der Eltern haben ein 50 %iges Risiko selbst asymptomatische Träger zu sein. 25 % der Geschwister von Betroffenen sind selbst nicht betroffen und auch keine Träger. Es ist nicht bekannt, dass CS-Betroffene vom Typ I oder II selbst Kinder bekommen haben. Die Kinder von CS-Betroffenen vom Typ III sind zu 100 % heterozygote Träger für eine pathogene Variante des CSA/ERCC8- oder CSB/ERCC6-Gens.[17]
Forschung
Die Erkrankung und ihre Ursachen hat in den letzten Jahrzehnten in Fachkreisen an Aufmerksamkeit gewonnen. In Deutschland wird aktuell daran geforscht, ob die Gabe künstlich hergestellter Chaperone, den Krankheitsverlauf positiv beeinflussen kann.[25][26] Bisher wurde dies jedoch erst an Mäusen getestet und es gibt keine klinischen Studien am Menschen (Stand 2020).
An der Universität Köln wurden die Auswirkungen des für CS verantwortlichen Gendefekts am Modell von Fadenwürmern nachgestellt, um die zugrundeliegenden Mechanismen besser zu verstehen und mögliche pharmakologische Behandlungen zu untersuchen.[27][28]
Weitere Informationen
- Amy and friends ist ein britisches Netzwerk für Betroffene, deren Familien und Angehörige von Cockayne-Syndrome und Trichothiodystrophy.[29]
- Share and Care Cockayne Syndrome Network ist ein US-amerikanisches Netzwerk für Betroffene, deren Familien und Angehörige.[30]
Beide Netzwerke veranstalten regelmäßig Konferenzen für Familien, deren Angehörige und behandelnde Fachleute mit internationalen Gästen.
Auf der Homepage der gemeinnützigen Organisation National Initiative for Cockayne Syndrome wurde im Januar 2021 je ein Handbuch für Eltern und Pflegende von Kindern mit CS, sowie ein Handbuch für Gesundheitsdienstleiste in englischer Sprache veröffentlicht.[31]
Differentialdiagnose
Abzugrenzen ist unter anderem das Flynn-Aird-Syndrom.
Literatur
- G. Hirschfeld, M. Berneburg, C. Arnim u. a.: Progeroid-Syndrome: Klinik und Molekularbiologie der vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 104, Nummer 6, 2007, S. A-346 / B-305 / C-292
- X. Yuan, W. Feng u. a.: Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. In: Molecular cell. Band 27, Nummer 4, August 2007, S. 585–595, ISSN 1097-2765. doi:10.1016/j.molcel.2007.06.021. PMID 17707230.
- C. Ovaert, A. Cano, B. Chabrol: Aortic dilatation in Cockayne syndrome. In: American journal of medical genetics. Part A. Band 143A, Nummer 21, November 2007, S. 2604–2606, ISSN 1552-4825. doi:10.1002/ajmg.a.31986. PMID 17935247.
Weblinks
- Cockayne-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Cockayne-Syndrom Typ A. In: Online Mendelian Inheritance in Man. (englisch)
- Cockayne-Syndrom Typ B. In: Online Mendelian Inheritance in Man. (englisch)
- Cockayne-Syndrom Typ C. In: Online Mendelian Inheritance in Man. (englisch)
- cockaynesyndrome.org
- Genes and Disease: Artikelsammlung
- Long-range four-stranded DNA structures found to play a role in rare ageing disease, auf: EurekAlert! vom 6. Dezember 2021 – über CSB und G-Quadruplex (englisch)
Einzelnachweise
- Martha A. Nance, Susan A. Berry: Cockayne syndrome: Review of 140 cases. In: American Journal of Medical Genetics. Band 42, Nr. 1, 1992, ISSN 1096-8628, S. 68–84, doi:10.1002/ajmg.1320420115.
- Brian T. Wilson, Zornitza Stark, Ruth E. Sutton, Sumita Danda, Alka V. Ekbote: The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. In: Genetics in Medicine. Band 18, Nr. 5, Mai 2016, ISSN 1530-0366, S. 483–493, doi:10.1038/gim.2015.110, PMID 26204423.
- Vincent Laugel: Cockayne syndrome: The expanding clinical and mutational spectrum. In: Mechanisms of Ageing and Development (= Special Issue on the segmental progeria Cockayne syndrome). Band 134, Nr. 5, 1. Mai 2013, ISSN 0047-6374, S. 161–170, doi:10.1016/j.mad.2013.02.006.
- L. Pasquier, V. Laugel, L. Lazaro, H. Dollfus, H. Journel: Wide clinical variability among 13 new Cockayne syndrome cases confirmed by biochemical assays. In: Archives of Disease in Childhood. Band 91, Nr. 2, 1. Februar 2006, ISSN 0003-9888, S. 178–182, doi:10.1136/adc.2005.080473, PMID 16428367.
- Valerie Natale: A comprehensive description of the severity groups in Cockayne syndrome. In: American Journal of Medical Genetics Part A. Band 155, Nr. 5, 2011, ISSN 1552-4833, S. 1081–1095, doi:10.1002/ajmg.a.33933.
- E. A. Cockayne: Dwarfism with retinal atrophy and deafness. In: Archives of Disease in Childhood. Band 11, Nr. 61, 1. Februar 1936, ISSN 0003-9888, S. 1–8, doi:10.1136/adc.11.61.1, PMID 21032019.
- E. A. Cockayne: Dwarfism with Retinal Atrophy and Deafness. In: Archives of Disease in Childhood. Band 21, Nr. 105, März 1946, ISSN 0003-9888, S. 52–54, PMC 1987981 (freier Volltext).
- Catherine A. Neill, Mary M. Dingwall: A Syndrome Resembling Progeria: A Review of Two Cases. In: Archives of Disease in Childhood. Band 25, Nr. 123, 1. September 1950, ISSN 0003-9888, S. 213–223, doi:10.1136/adc.25.123.213, PMID 14783428.
- Debora Ferri, Donata Orioli, Elena Botta: Heterogeneity and overlaps in nucleotide excision repair disorders. In: Clinical Genetics. Band 97, Nr. 1, 2020, ISSN 1399-0004, S. 12–24, doi:10.1111/cge.13545.
- Alain J. van Gool, Gijsbertus T. J. van der Horst, Elisabetta Citterio, Jan H. J. Hoeijmakers: Cockayne syndrome: defective repair of transcription? In: The EMBO Journal. Band 16, Nr. 14, 15. Juli 1997, ISSN 0261-4189, S. 4155–4162, doi:10.1093/emboj/16.14.4155, PMID 9250659.
- Ajoy C. Karikkineth, Morten Scheibye-Knudsen, Elayne Fivenson, Deborah L. Croteau, Vilhelm A. Bohr: Cockayne syndrome: Clinical features, model systems and pathways. In: Ageing Research Reviews (= Monogenic Accelerated Aging Disorders with Perturbations to Normal DNA and Chromosome Function). Band 33, 1. Januar 2017, ISSN 1568-1637, S. 3–17, doi:10.1016/j.arr.2016.08.002, PMID 27507608.
- Morten Scheibye-Knudsen, Deborah L. Croteau, Vilhelm A. Bohr: Mitochondrial deficiency in Cockayne syndrome. In: Mechanisms of Ageing and Development (= Special Issue on the segmental progeria Cockayne syndrome). Band 134, Nr. 5, 1. Mai 2013, ISSN 0047-6374, S. 275–283, doi:10.1016/j.mad.2013.02.007, PMID 23435289.
- Agnès Bloch-Zupan, Morgan Rousseaux, Virginie Laugel, Matthieu Schmittbuhl, Rémy Mathis: A possible cranio-oro-facial phenotype in Cockayne syndrome. In: Orphanet Journal of Rare Diseases. Band 8, Nr. 1, 14. Januar 2013, ISSN 1750-1172, S. 9, doi:10.1186/1750-1172-8-9, PMID 23311583.
- Sarah Baer, Nicolas Tuzin, Peter B. Kang, Shehla Mohammed, Masaya Kubota: Growth charts in Cockayne syndrome type 1 and type 2. In: European Journal of Medical Genetics. Band 64, Nr. 1, Januar 2021, S. 104105, doi:10.1016/j.ejmg.2020.104105 (elsevier.com [abgerufen am 4. Februar 2021]).
- V. Laugel, C. Dalloz, M. Durand, F. Sauvanaud, U. Kristensen: Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. In: Human Mutation. Band 31, Nr. 2, 2010, ISSN 1098-1004, S. 113–126, doi:10.1002/humu.21154.
- Metabolic Study of Cockayne Syndrome (METABO-CS). In: U.S. National Library of Medicine. 6. Februar 2017, abgerufen am 31. Mai 2020.
- Vincent Laugel: Cockayne Syndrome. In: GeneReviews®. University of Washington, Seattle, Seattle (WA) 1993, PMID 20301516 (nih.gov [abgerufen am 31. Mai 2020]).
- Brian T. Wilson, Andrew Strong, Sean O’Kelly, Jennifer Munkley, Zornitza Stark: Metronidazole Toxicity in Cockayne Syndrome: A Case Series. In: Pediatrics. Band 136, Nr. 3, 1. September 2015, ISSN 0031-4005, S. e706–e708, doi:10.1542/peds.2015-0531, PMID 26304821.
- Isabelle Rapin, Karen Weidenheim, Yelena Lindenbaum, Pearl Rosenbaum, Saumil N. Merchant: Cockayne Syndrome in Adults: Review With Clinical and Pathologic Study of a New Case. In: Journal of Child Neurology. Band 21, Nr. 11, November 2006, ISSN 0883-0738, S. 991–1006, doi:10.1177/08830738060210110101.
- Fluid overload | Share and Care Network. Abgerufen am 31. Mai 2020 (amerikanisches Englisch).
- Masaya Kubota, Sayaka Ohta, Aki Ando, Akiko Koyama, Hiroshi Terashima: Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. In: Pediatrics International. Band 57, Nr. 3, 2015, ISSN 1442-200X, S. 339–347, doi:10.1111/ped.12635.
- Wim J. Kleijer, Vincent Laugel, Mark Berneburg, Tiziana Nardo, Heather Fawcett: Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. In: DNA Repair. Band 7, Nr. 5, 3. Mai 2008, ISSN 1568-7864, S. 744–750, doi:10.1016/j.dnarep.2008.01.014.
- Morad Khayat, Hagar Hardouf, Joel Zlotogora, Stavit Allon Shalev: High carriers frequency of an apparently ancient founder mutation p.Tyr322X in the ERCC8 gene responsible for Cockayne syndrome among Christian Arabs in Northern Israel. In: American Journal of Medical Genetics Part A. 152A, Nr. 12, Dezember 2010, S. 3091–3094, doi:10.1002/ajmg.a.33746 (wiley.com [abgerufen am 9. Februar 2021]).
- Tzipora C. Falik‐Zaccai, Meital Laskar, Nechama Kfir, Wael Nasser, Hanoch Slor: Cockayne syndrome type II in a Druze isolate in Northern Israel in association with an insertion mutation in ERCC6. In: American Journal of Medical Genetics Part A. 146A, Nr. 11, 2008, ISSN 1552-4833, S. 1423–1429, doi:10.1002/ajmg.a.32309 (wiley.com [abgerufen am 9. Februar 2021]).
- Marius Costel Alupei, Pallab Maity, Philipp Ralf Esser, Ioanna Krikki, Francesca Tuorto: Loss of Proteostasis Is a Pathomechanism in Cockayne Syndrome. In: Cell Reports. Band 23, Nr. 6, 8. Mai 2018, ISSN 2211-1247, S. 1612–1619, doi:10.1016/j.celrep.2018.04.041, PMID 29742419.
- Annika Bingmann: Cockayne-Syndrom: Altern wie im Zeitraffer Neuer Therapieansatz für „kindliche Greise“. 8. Mai 2018, abgerufen am 31. Mai 2020.
- Björn Schumacher erhält Eva Luise Köhler Forschungspreis 2019: Seltene Erkrankungen geben Einblick in Geheimnis des Alterns. 18. Februar 2019, abgerufen am 31. Mai 2020.
- Was beim Cockayne Syndrom passiert. 15. Februar 2019, abgerufen am 31. Mai 2020.
- amyandfriends.org
- cockaynesyndrome.org
- National Initiative for Cockayne Syndrome (NICS). Abgerufen am 4. Februar 2021.