Retinopathia pigmentosa
Die Bezeichnung Retinopathia pigmentosa oder Retinitis pigmentosa (RP) beschreibt eine durch Vererbung oder spontane Mutation entstehende Netzhautdegeneration, bei der die Photorezeptoren zerstört werden. Man spricht von Pseudoretinitis pigmentosa (oder Phänokopie), wenn nicht erbliche Erkrankungen Symptome der Retinopathia pigmentosa zeigen, etwa toxisch bedingt (beispielsweise durch Thioridazin, Chloroquin).
| Klassifikation nach ICD-10 | |
|---|---|
| H35.5 | Hereditäre Netzhautdystrophie |
| ICD-10 online (WHO-Version 2019) | |
Der Name Retinitis pigmentosa wurde von dem Niederländer Frans Donders im Jahr 1855 geprägt.[1] Da es sich hierbei jedoch nicht primär um eine Entzündung (-itis) handelt, wurde die Krankheit in Retinopathia pigmentosa umbenannt. Die ursprüngliche Benennung wird allerdings immer noch synonym und sogar häufiger verwendet als die neue Bezeichnung.
Die entsprechende Erkrankung bei Tieren wird in der Veterinärmedizin als progressive Retinaatrophie (PRA) bezeichnet.
Epidemiologie
Weltweit sind etwa drei Millionen Menschen – in Deutschland etwa 30.000 bis 40.000 – von einer der verschiedenen Formen der Retinopathia pigmentosa (auch genannt Patermann-Syndrom) betroffen. Bedingt durch den schleppenden Verlauf der Erkrankung dürfte die Dunkelziffer noch höher sein. Die Prävalenz liegt somit bei einem Fall pro 3000 bis 7000 Einwohnern.[2]
Die Erkrankung tritt meistens im Jugendalter oder in den mittleren Lebensjahren mit den ersten Merkmalen (Nachtblindheit) ein; die Sehkraft lässt allmählich nach. Der gesamte Prozess zunehmender Sehbehinderung verläuft schleichend und erstreckt sich beim Betroffenen meistens über Jahrzehnte hinweg. Diese Entwicklung ist so auch mit einer starken psychischen Belastung verbunden. Bei ca. der Hälfte aller RP-Patienten entwickelt sich im Erwachsenenalter eine Linsentrübung, der graue Star.[3]
Symptome und Verlauf
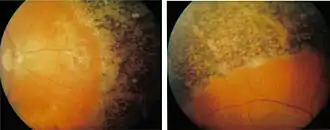
Es treten in der Regel folgende Symptome auf:
- Nachtblindheit (erste Anhaltspunkte)
- Tunnelblick, konzentrische Einschränkung des Gesichtsfeldes (frühe Anzeichen)
- schlechte Adaptation des Auges an sich ändernde Lichtbedingungen
- Blendempfindlichkeit
- Störung im Kontrastsehen
- Beeinträchtigung des Farbsehens
- allmählicher Verlust der Sehfähigkeit bis zur kompletten Erblindung
Im Verlauf kommt es typischerweise erst zu Nachtblindheit, Visusabfall und dann zu einer langsamen Einschränkung des Gesichtsfeldes bis hin zu einem sich immer mehr verengenden Tunnelblick. Die Erkrankung führt in einem späteren Stadium in der Regel zur Blindheit. Wegen der Nachtblindheit und des Tunnelblicks können sich die Patienten kaum mehr ohne Einsatz blindenspezifischer Orientierungsstrategien und eines Langstocks alleine sicher fortbewegen. Der zeitliche Verlauf dieser Abfolge variiert dabei, abhängig vom jeweiligen RP-auslösenden Gendefekt.[4]
Das Absterben der Photorezeptoren, zuerst der Stäbchen, vollzieht sich in der Regel von der Peripherie (dem Rand des Gesichtsfeldes) zur Makula (dem Zentrum des Gesichtsfeldes) hin. Donders beschrieb besonders die knochenkörperchenähnlichen Pigmenteinlagerungen und Gefäßverengungen im Auge, welche der Retinopathia pigmentosa den Namen gaben. Diese Veränderungen treten aber in der Regel sekundär zur Degeneration der Photorezeptoren auf.
Genetik
Über 71 Gene wurden inzwischen identifiziert, deren Defekt Retinopathia pigmentosa auslösen kann.[5] Die meisten bislang identifizierten Gene folgen einem monogenetischem Erbgang, d. h. der Defekt von nur einem Gen verursacht bereits die Erkrankung und nicht von mehreren Genen gleichzeitig. Die Erkrankung wird sowohl durch autosomal dominante, autosomal rezessive und auch gonosomale Erbfaktoren (im Wesentlichen das X-Chromosom) ausgelöst.[2][3]
Syndrome
Ungefähr 25 % der betroffenen Patienten leiden an einer assoziierten Retinopathia pigmentosa. Bei der assoziierten RP weisen neben dem Auge auch andere Organe des Körpers Krankheitssymptome auf, das heißt, es liegt ein Syndrom vor.[2] Einige solcher mit Retinopathia pigmentosa oftmals zusammen auftretenden Symptome sind Hörstörungen, Lähmungen und Gehstörungen, Herzrhythmusstörungen, Muskelschwäche, geistige Entwicklungsstörungen u. a. Auch hier sind Gendefekte die Ursache. Die bekanntesten Syndrome sind:
- das Usher-Syndrom,
- das Bardet-Biedl-Syndrom,
- das Refsum-Syndrom,
- das NBIA-Syndrom,
- das Alport-Syndrom
- das Saldino-Mainzer-Syndrom.
Diagnose
Bereits in früher Kindheit kann eine Retinopathia pigmentosa über ein Elektroretinogramm diagnostiziert werden. Weitere Diagnosemöglichkeiten bieten Sehtests zur Nachtblindheit beim Augenarzt. Bei Syndromen geben die weiteren Symptome Hinweise auf die genaue Erkrankung (wie Hörstörungen oder Blutwerte). Die Ermittlung des genauen Gendefekts ermöglicht erst eine DNA-Analyse. Noch in der Entwicklung befinden sich DNA- und Protein-Chips, welche eine schnellere Diagnose ermöglichen sollen. Dies kann auch zur genetischen Familienberatung beitragen.
Abzugrenzen ist unter anderem die Choroideremie.
Behandlung
Es gibt derzeit keine Behandlung, die das Fortschreiten von Retinopathia pigmentosa verhindern oder die Krankheit heilen kann. Allerdings existieren Studien, wonach die Einnahme von Vitamin A oder Anti-Vascular Endothelial Growth Factor den Verlauf verlangsamen soll.[6] Nachfolgestudien sehen eine mögliche Wirksamkeit von Vitamin A jedoch nur bei bestimmten Genmutationen gegeben.[7] Eine weitere Studie zeigt, dass der Krankheitsverlauf durch hyperbare Sauerstofftherapie verlangsamt werden kann.[8] Eine Ausnahme bilden Sonderformen wie das Refsum-Syndrom, ein Stoffwechseldefekt, bei dem eine phytansäurearme Spezialdiät oder, falls diese nicht ausreicht, regelmäßige Lipidapherese die Retinopathia pigmentosa zum Stillstand bringen kann.[9][10]
Noch in der Erforschung befinden sich gentherapeutische Ansätze, bei denen defekte Gene in der Retina ersetzt werden könnten, oder Stammzelltherapien, bei denen die degenerierte Retina repariert werden soll.[6][11][12][13] In der Entwicklung sind auch sogenannte Retina-Implantate, bei denen Mikrosystemtechnik als Prothese die Funktionen der defekten Retina ersetzen soll.[14]
Kritisch gesehen werden Behandlungsansätze wie die Akupunktur[15] oder andere Therapien, deren Wirksamkeit nicht durch wissenschaftliche Studien abgesichert ist, wie etwa die sogenannte Kuba-Therapie.[16]
Der 2020 mit dem Körber-Preis ausgezeichnete Mediziner Botond Roska untersucht, wie geschädigte Netzhäute mit Gentherapie geheilt werden können. 2021 gelang es einem internationalen Forschungsteam erstmals, einem durch Retinitis pigmentosa erblindeten 58-jährigen Patienten mit einer optogenetischen Therapie partiell das Sehen wieder zu ermöglichen.[17]
Literatur
- C. Hamel: Retinitis pigmentosa. In: Orphanet Journal of Rare Diseases. 1, 2006, S. 40. doi:10.1186/1750-1172-1-40
- Aleksandra Polosukhina, Jeffrey Litt u. a.: Photochemical Restoration of Visual Responses in Blind Mice. In: Neuron. 75, 2012, S. 271–282, doi:10.1016/j.neuron.2012.05.022.
Weblinks
- Pro Retina Deutschland e. V. Pro Retina Deutschland ist eine Selbsthilfevereinigung von Menschen mit Netzhautdegenerationen.
- Leitlinie Nr. 25 – Hereditäre Netzhaut-, Aderhaut- oder Sehbahn-Erkrankungen. (PDF) Berufsverband der Augenärzte Deutschlands e. V. Deutsche Ophthalmologische Gesellschaft e. V.
Einzelnachweise
- F. C. Donders: Beiträge zur pathologischen Anatomie des Auges. In: Arch für Ophthalmol. 1855-1, S. 106–118.
- S. P. Daiger u. a.: Perspective on genes and mutations causing retinitis pigmentosa. In: Arch Ophthalmol. 125(2), Feb 2007, S. 151–158. PMID 17296890
- D. T. Hartong u. a.: Retinitis pigmentosa. In: Lancet. 368(9549), 18. Nov 2006, S. 1795–1809. PMID 17113430
- M. A. Sandberg u. a.: Disease Course in Patients with Autosomal Recessive Retinitis Pigmentosa due to the USH2A Gene. In: Invest Ophthalmol Vis Sci. 18. Jul 2008, S. 5532–5539. PMID 18641288.
- José-Alain Sahel, Elise Boulanger-Scemama, Chloé Pagot, Angelo Arleo, Francesco Galluppi, Joseph N. Martel, Simona Degli Esposti, Alexandre Delaux, Jean-Baptiste de Saint Aubert, Caroline de Montleau, Emmanuel Gutman, Isabelle Audo, Jens Duebel, Serge Picaud, Deniz Dalkara, Laure Blouin, Magali Taiel & Botond Roska: Partial recovery of visual function in a blind patient after optogenetic therapy. In: Nature Medicine. 24. Mai 2021. doi:10.1038/s41591-021-01351-4.
- P. Goodwin: Hereditary retinal disease. In: Curr Opin Ophthalmol. 19, (3), Mai 2008, S. 255–262. PMID 18408503
- U. Kellner: Stellungnahme des Arbeitskreises Klinische Fragen (AKF) des Wissenschaftlich-Medizinischen Beirats der Pro Retina Deutschland e.V. (Stand: 11.03.2016). Stellungnahme zur Gabe von Vitamin A bei erblichen Netzhautdystrophien, abgerufen am 31. August 2020.
- Enzo Maria Vingolo et al.: Slowing the degenerative process, long lasting effect of hyperbaric oxygen therapy in retinitis pigmentosa, 2008, abgerufen am 31. August 2020
- K. Rüether, E. Baldwin, M. Casteels, M. D. Feher, M. Horn, S. Kuranoff, B. P. Leroy, R. J. Wanders, A. S. Wierzbicki: Adult Refsum disease: a form of tapetoretinal dystrophy accessible to therapy. In: Surv Ophthalmol. 55 (6), 2010, S. 531–538. PMID 20850855.
- E. J. Baldwin, F. B. Gibberd, C. Harley, M. C. Sidey, M. D. Feher, A. S. Wierzbicki: The effectiveness of long-term dietary therapy in the treatment of adult Refsum disease. In: J Neurol Neurosurg Psychiatry. 81 (9), 2010, S. 954–957. PMID 20547622.
- I. Mooney, J. LaMotte: A review of the potential to restore vision with stem cells. In: Clin Exp Optom. 91 (1), Jan 2008, S. 78–84. PMID 18045253
- William A. Beltran, Artur V. Cideciyan u. a.: Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. In: Proceedings of the National Academy of Sciences. 112, 2015, S. E5844–E5853, doi:10.1073/pnas.1509914112.
- A. G. Bassuk u. a.: Precision Medicine: Genetic Repair of Retinitis Pigmentosa in Patient-Derived Stem Cells. In: Sci. Rep. 6, 2016, S. 19969. doi:10.1038/srep19969
- N. Alteheld u. a.: Towards the bionic eye--the retina implant: surgical, ophthalmological and histopathological perspectives. In: Acta Neurochirurgica Suppl. 97 (Pt 2), 2007, S. 487–493. PMID 17691339.
- Stellungnahme des Arbeitskreises Klinische Fragen des Wissenschaftlich-Medizinischen Beirats der PRO RETINA Deutschland e. V. zur Akupunktur bei Retinitis pigmentosa und anderen erblichen Netzhauterkrankungen, Stand: 01.07.2014, abgerufen am 31. August 2020.
- Stellungnahme der gemeinsamen Kommission von DOG und BVA zur Evaluation alternativer/komplementärer Angebote in der Augenheilkunde zur Kuba-Therapie“ bei tapetoretinalen Degenerationen (Retinitis Pigmentosa) und aktuellen Marketing-Maßnahmen von Kuba-Therapies Ltd. In Deutschland, Stand 31.03.2009, abgerufen am 31. August 2020.
- Optogenetische Gentherapie lässt Erblindeten partiell wieder Sehen. 24. Mai 2021, abgerufen am 28. Mai 2021 (deutsch).