Progerie
Progerie, auch Progeria und vorzeitige Alterung (hergeleitet von altgriechisch πρό (pró) – vor und τὸ γῆρας (γήρας), -ως bzw. γῆρος, -ους (gäras) – Alterung, Seneszenz (von lat. senescere – altern)), im engeren Sinn das Hutchinson-Gilford-Progerie-Syndrom (HGPS), auch Progeria infantilis genannt, gehört zu den segmental progeroiden Syndromen (SPS; seltene Erkrankungen mit Zeichen einer vorzeitigen Alterung in mehr als einem Organ oder Gewebe) oder Progerie-Syndromen[1] und ist ein Krankheitszeichen verschiedener Erbkrankheiten, die bei Kindern mit einem überschnellen Altern einhergehen (Akrogerie, Bloom-Syndrom, Cockayne-Syndrom, Geroderma osteodysplastica, Ogden-Syndrom, Xeroderma pigmentosum).
| Klassifikation nach ICD-10 | |
|---|---|
| E34.8 | Sonstige näher bezeichnete endokrine Störungen |
| ICD-10 online (WHO-Version 2019) | |
Das Krankheitsbild wurde erstmals 1886 von Jonathan Hutchinson und Hastings Gilford beschrieben. Auffälligstes Merkmal ist eine vorzeitige Vergreisung der betroffenen Kinder.
Daneben gibt es auch die Form der Progeria adultorum (Werner-Syndrom), die bei Erwachsenen etwa ab der Lebensmitte zu einem beschleunigten Altern führt.
Krankheitsanzeichen
Die von dieser Erkrankung betroffenen Kinder werden ohne Auffälligkeiten geboren und entwickeln erste Symptome im Alter von sechs bis zwölf Monaten. Folgende Symptome können auftreten:[2]
- Kleinwuchs
- Haarausfall (Alopezie)
- Verlust des Unterhaut-Fettgewebes, Folge: Pergamenthaut
- Nagelhypoplasie (fehlgebildete Nägel)
- kraniofaziale Fehlbildung mit u. a. Rückverlagerung des Unterkiefers im Verhältnis zur Schädelbasis (Mikrognathie)
- Osteolyse (Knochenschwund)
- Atherosklerose („Arterienverkalkung“); primär für die verkürzte Lebenserwartung verantwortlich.[2]
Häufigkeit und Krankheitsverlauf
Die Betroffenen altern fünf- bis zehnmal schneller als Menschen ohne diese Krankheit. Die häufigsten Todesursachen sind Herzinfarkt und Schlaganfall, die bereits im Kindes- oder Jugendalter auftreten. Viele Phänomene des normalen Alterns treten bei Kindern mit HGPS jedoch nicht auf; so ist das Tumorrisiko nicht relevant gesteigert, und auch neurodegenerative Erkrankungen wie die Alzheimer-Krankheit treten nicht gehäuft auf. HGPS ist damit also keine exakte Kopie des üblichen Alterns. Die Lebenserwartung liegt bei vierzehn Jahren. Die Prävalenz wird auf 1:4.000.000 geschätzt. Weltweit sollen etwa 200–250 Kinder mit HGPS leben. Bis 2013 wurden ca. 100 Fälle identifiziert.[3]
Da die molekularen Mechanismen der Regulation von Altersprozessen nur unvollständig bekannt sind, besteht keine Möglichkeit zur Vorbeugung oder zur Therapie mittels medikamentöser Beeinflussung derzeit erforschter molekularer Vorgänge bei altersassoziierten Erkrankungen wie den segmental progeroiden Syndromen.[4]
Ursache
Eine Ursache für Progerie ist eine Punktmutation c.794 A→G (N265S) (Chromosom 1 Genlocus p34.2) im ZMPSTE24-Gen, welches das Enzym CAAX-Prenylprotease bzw. Zink-Metallopeptidase-Ste-24-homolog codiert.[5] Dieses ist essentiell für die Bildung des Strukturproteins Lamin A, das ein wichtiger Bestandteil der inneren Zellkernmembran ist. Mutiertes ZMPSTE24 kann Prälamin A nicht notwendigerweise an der Aminosäure 647 trennen, um die Prenylierung zu eliminieren und das fertige, 646 AA (Aminosäuren) lange Lamin A herzustellen. In der Mehrzahl der Fälle von HGPS findet allerdings eine Mutation des Prälamin-Gens im Codon 608 auf Chromosom 1 Genlocus q23 selbst statt, die das Trinucleotid GGC in GGT ändert. Dies codiert zwar für die gleiche Aminosäure (Gly), jedoch wird durch die veränderte Basenreihung eine Spleißstelle (5'AC) in der entsprechenden prä-mRNA eingefügt. Es entsteht bei diesem fehlerhaften Spleißen eine um 150 Basen kürzere Prelamin-RNA und ein um 50 AA kürzeres Lamin A, das auch als Progerin bezeichnet wird. Die Schnittstelle zur Prozessierung zum Lamin A fehlt, und ZMPSTE24 kann Prälamin A nicht schneiden. Es bleibt ein zu kurzes prenyliertes Lamin bestehen. Seltener findet man eine Änderung des Codons 608 in AGT oder eine Mutation des Codon 145.
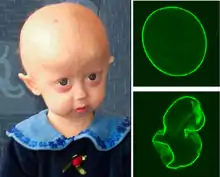
Lamin A (Strukturprotein der Kernmembran, das vom zwölf Exons enthaltenden Gen LMNA kodiert wird) ist Bestandteil einer Proteinkette, die an zahlreiche andere Proteine des Zellkerns und der Zellkernmembran sowie an Transkriptionsfaktoren und die DNA bindet. Es nimmt eine stabilisierende Funktion des Zellkerns sowie regulatorische Funktionen wahr. Unter anderem nimmt es an der Aktivierung von Genen teil. Die Vererbung der Hutchinson-Gilford-Progerie ist autosomal-dominant. Bereits ein defektes Allel reicht aus, um die Erkrankung zu verursachen. Dies liegt an der Kettenstruktur, die durch Lamin A gebildet wird. Bereits einige wenige defekte Lamin-A-Proteine führen zur Instabilität der gesamten Kette. Die Zellkerne der Menschen mit HGPS sind daher zum großen Teil deformiert.[6]
Da die Kinder in der Regel das reproduktionsfähige Alter nicht erreichen, ist eine Neuerkrankung praktisch immer eine Spontanmutation im Lamin-A-Gen (dominanter Letalfaktor). Wenige erbliche Fälle der HGPS sind beschrieben worden. Häufig haben diese Kinder jedoch nicht das typische Bild der Erkrankung, sondern Varianten mit weiteren klinischen Auffälligkeiten und einer anderen Lebenserwartung. Hier sind auch autosomal-rezessive Vererbungen beschrieben worden; teils durch andere Mutationen im Lamin-A-Gen, teils durch Mutationen in Genen, die an der Reifung (Prozessierung) eines Vorläuferproteins, des Prälamin A, zu Lamin A mitwirken.
Differentialdiagnose
Abzugrenzen sind die Mandibuloakrale Dysplasie, das Wiedemann-Rautenstrauch-Syndrom (ein kongenitales segmentales progeroides Syndrom durch Mutationen im Gen POLR3A[7]), das GAPO-Syndrom, CARASIL und die Akrogerie.
Wie das Hutchinson-Gilford-Progerie-Syndrom gehört auch das Mandibuläre Hypoplasie-, Taubheit-, progeroide-Merkmale- und Lipodystrophie-Syndrom (MDPL) zu den infantilen segmental progeroiden Syndromen. Die genetische Ursache des MDPL sind heterozygote Mutationen im Gen POLD1.[8]
Sonstiges
- Eine der Hauptfiguren der Otherland-Romane von Tad Williams, Orlando Gardiner, leidet an Progerie.
- Der südafrikanische Maler und Hip-Hop-DJ Leon Botha (1985–2011) litt ebenfalls an Progerie und starb daran. Er wurde 26 Jahre alt.
- Öffentliche Aufmerksamkeit erlangte die Krankheit in den USA durch Sam Berns und die von HBO ausgestrahlte Realityshow Life according to Sam.[9]
Literatur
- M. A. Merideth u. a.: Phenotype and Course of Hutchinson-Gilford Progeria Syndrome. In: New England Journal of Medicine. Nr. 358, 2008, S. 592–604 (Abstract).
- Michael Schophaus: Zu jung, um alt zu sein: Die Geschichte einer rätselhaften Krankheit. Goldmann, München 2004, ISBN 978-3-442-15279-7.
- Bruno Schrep: Warum mein Kind? In: Der Spiegel Nr. 38 vom 16. September 2002, S. 178ff.
- Hayley Okines u. a.: Old Before My Time: Hayley Okines' Life with Progeria. 2011, ISBN 978-1-908192-55-4.
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496, insbesondere (zum Hutchinson-Gilford-Progerie-Syndrom) S. 491–493.
Filme
- Jack (1996), US-amerikanisches Filmdrama von Francis Ford Coppola
- Sarahs kurzes Leben (2003/ORF) von Manfred Corrine
- Sabrina – Zu jung um alt zu sein (2000/ Schweizer Fernsehen) von Elsbeth Leisinger
- Paa – A very Rare Father-Son, Son-Father Story (2009) von R. Balki
- Bjorn, gewoon YOLO (2015/ZAPP, Niederlande), Dokumentation von Siham Raijoul[10]
- My philosophy for a happy life[11], TED-Talk von Sam Berns, einem an Progeria erkrankten Jugendlichen, englischsprachig
Weblinks
Einzelnachweise
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496.
- Hutchinson-Gilford-Syndrom Orphanet, abgerufen am 22. Mai 2021.
- F. Coppedè: The epidemiology of premature aging and associated comorbidities. In: Clinical interventions in aging. Band 8, 2013, S. 1023–1032, doi:10.2147/CIA.S37213, PMID 24019745, PMC 3760297 (freier Volltext) (Review).
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496.
- S. Shackleton u. a.: Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. In: J Med Genet 2005,42;e36. doi:10.1136/jmg.2004.029751.
- M. Eriksson u. a.: Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. In: Nature 423, 2003, S. 293–298. PMID 12714972
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496, hier: S. 490 f.
- Davor Lessel, Christian Kubisch: Genetisch bedingte Syndrome mit Zeichen einer vorzeitigen Alterung. In: Deutsches Ärzteblatt. Band 116, Heft 29 f., 22. Juli 2019, S. 489–496, hier: S. 491 f.
- Rat eines Sterbenden – „Verpasst nie eine Party“ In: welt.de, Abgerufen am 24. Mai 2019
- http://jeugdjournaal.nl/item/767469-bjorn-gewoon-yolo.html die Dokumentation Bjorn, gewoon YOLO auf jeugdjournaal.nl (niederl.)
- Meine Philosophie für ein glückliches Leben | Sam Berns | TEDxMidAtlantic In: YouTube, Abgerufen am 24. Mai 2019