DNA-Reparatur
Durch Mechanismen der DNA-Reparatur können Zellen schädliche Veränderungen ihrer DNA-Struktur beseitigen. Solche Schäden in der DNA können spontan im Verlauf der DNA-Replikation oder durch die Einwirkung mutagener Substanzen, extremer Wärme oder ionisierender Strahlung verursacht werden.
| Übergeordnet |
| DNA-Metabolismus |
| Untergeordnet |
| Einzelstrangbruch-Reparatur Doppelstrangbruch-Reparatur Postreplikations-Reparatur Virale DNA-Reparatur mitochondrielle DNA-Reparatur Pyrimidindimer-Reparatur Basen-Exzisionsreparatur Nukleotid-Exzisionsreparatur |
| Gene Ontology |
|---|
| QuickGO |
DNA-Schäden können dazu führen, dass die Replikation der DNA für die Mitose falsch erfolgt, Proteine nicht mehr bzw. falsch synthetisiert oder wichtige Chromosomenbereiche nach Doppelstrangbrüchen abgespalten werden.
Bringen die komplexen Reparaturmechanismen der Zelle keinen Erfolg, so sammeln sich in wachsenden und ruhenden somatischen Zellen so viele Fehler an, dass die normalen Zellfunktionen gestört sind. In einer Keimzelle wären die Tochterzellen nicht mehr lebensfähig, was zu einer Inaktivierung der Zelllinie führt: die Zelle bzw. die zweite bis dritte nachfolgende Generation verliert ihre Teilungsfähigkeit und stirbt. Im Zuge der Zellzykluskontrolle können Kontrollproteine eine Zelle bzw. deren DNA als defekt erkennen und einen Zyklusarrest (G0-Phase) oder den programmierten Zelltod (Apoptose) einleiten.[1]
Einzelne DNA-Reparaturenzyme konnten inzwischen mit PAL-Mikroskopie bei ihrer Arbeit in einem Bakterium verfolgt und die entsprechenden Parameter bestimmt werden. So dauert beispielsweise in E. coli eine Basenexzisionsreparatur gut zwei Sekunden.[2]
Ursachen von DNA-Schäden
Mögliche Ursachen sind Stoffwechselvorgänge, chemische Substanzen oder Ionisierende Strahlung, wie zum Beispiel UV-Strahlung, Elektronen oder Protonen.
Sowohl DNA-Schäden als auch Fehler bei der Replikation und anderen zellulären Prozessen können zu Mutationen führen. Da für Mutationen eigene Reparaturmechanismen existieren, können sie hier ähnlich wie DNA-Schäden betrachtet werden.
Stoffwechselvorgänge
Eine Zelle ist ein System im Fließgleichgewicht. Sie nimmt fortwährend Moleküle auf, verarbeitet sie, synthetisiert benötigte Stoffe, und gibt wiederum bestimmte Stoffe an die Umgebung ab. Beim normalen zellulären Metabolismus können reaktive Sauerstoffspezien (ROS, unter anderem Sauerstoffradikale) entstehen, welche ein signifikantes Ausmaß an oxidativen Schaden anrichten. Am häufigsten sind dies Basenschäden und Einzelstrangbrüche, weniger als 0,5 % sind Doppelstrangbrüche, welche auch noch relativ gleichförmig über die DNA verteilt sind. Die Wahrscheinlichkeit endogen induzierter Schadenscluster und damit – schwierig zu reparierender – gehäufter Läsionen (complex lesions), wie sie sonst durch die nicht-homogene Energieabgabe ionisierender Strahlen auftreten, ist sehr gering. Eine zu hohe Protonendichte und/oder zu hohe Temperatur kann Depurinierungen oder Depyrimidierung auslösen.
UV-Strahlung
Durch die UV-Strahlung kann es zu direkten Veränderungen (Mutationen) der DNA kommen,[3] wobei diese insbesondere UV-C-FUV-Strahlung absorbiert. Einzelsträngige DNA zeigt ihr Absorptionsmaximum bei 260 nm. Sowohl UV-B als auch UV-A können indirekt die DNA durch die Entstehung von reaktiven Sauerstoffradikalen schädigen, die die Entstehung von Oxidativen DNA-Läsionen bewirken, die wiederum zu Mutationen führen. Diese sind vermutlich für die Entstehung von UV-A-induzierten Tumoren verantwortlich.[4]
Arten von DNA-Schäden
- Basenmodifikationen
- Pyrimidindimere in der Regel 6–4-Photoprodukte (6-4PPs) oder Cyclobutan-Pyrimidindimere (CPDs)
- oxidierte Basen beispielsweise 8-Oxo-7,8-Dihydroguanin (8-oxoG) oder 8-Oxo-7,8-Dihydroadenin (8-oxoA)
- alkylierte Basen (z. B. Basenmethylierungen)
- andere Bulky lesions (sperrige Basenveränderungen)
- Basenfehlpaarungen durch fehlerhafte Replikation (Mismatch)
- Basenverlust – Apurinierungen oder Apyrimidierungen (AP sites)
- Veränderungen des Zuckergerüsts
- DNA-Protein-Vernetzungen (DNA protein crosslinks)
- DNA-DNA-Verknüpfungen (DNA crosslinks)
- Einzelstrangbrüche (ss breaks)
- Doppelstrangbrüche (ds breaks)
Die Behandlung mit einem Gray Röntgenstrahlung erzeugt pro Zelle etwa[5]
- 1000–2000 Basenmodifikationen
- 500–1000 Einzelstrangbrüche
- 800–1600 Veränderungen des Zuckergerüsts
- 150 DNA-Protein-Vernetzungen
- 50 Doppelstrangbrüche
Reparatur von DNA-Schäden
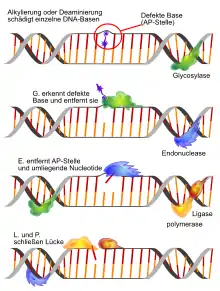
Für die unterschiedlichen Arten von DNA-Schäden existieren unterschiedliche, spezialisierte Reparaturmechanismen. Einige Mechanismen sind beispielsweise auf die Reparatur von Schäden im DNA-Einzelstrang, andere auf die Reparatur von DNA-Doppelstrangbrüchen spezialisiert.
Unterschiede bestehen auch zwischen Prokaryonten und Eukaryonten, die unterschiedliche DNA-Polymerasen besitzen.
Basenexzisionsreparatur (BER)
Bei der Basenexzisionsreparatur werden Fehler in Form oxidierter, alkylierter oder desaminierter einzelner Basen behoben. Dabei werden Schäden an den Basen durch eine jeweils spezifische DNA-Glykosylase erkannt und herausgeschnitten (Exzision). Diese wandert entlang der kleinen Furche und klappt die einzelnen Basen in ihr katalytisches Zentrum. Eine beschädigte Base wird von der DNA-Glykosylase entfernt, danach wird durch eine AP-Endonuklease (Apurinische/apyrimidinische-Endonuklease) ein Einzelstrangbruch im Zucker-Phosphat-Rückgrat eingeführt. Eine DNA-Polymerase synthetisiert abhängig von der komplementären Base auf dem fehlerfreien Strang die korrekte Base. Es existieren hier zwei Varianten der BER: short patch repair (eine einzelne Base wird ersetzt) und long patch repair (2-20 Nukleotide werden ersetzt). Beim Menschen ist die DNA-Polymerase β (Pol β) die hauptverantwortliche Polymerase. Eine DNA-Ligase verknüpft die neue Base im DNA-Strang, womit der Fehler korrigiert ist.
Nukleotidexzisionsreparatur (NER)
Im Unterschied zur Basenexzisionsreparatur werden von der Nukleotidexzisionsreparatur (NER) vor allem sogenannte „bulky lesions“ erkannt, also Stellen, die eine Art „Buckel“ im DNA-Molekül erzeugen und dadurch die Helixstruktur stören. Es kann sich dabei um Pyrimidindimere und 6,4 Photoprodukte handeln, welche durch UV-Strahlung erzeugt werden.
Die Nukleotidexzisionsreparatur gliedert sich in die Schadenserkennung, das Einschneiden, das Herausschneiden eines 25-30 Basen langen DNA-Abschnitts, die Neusynthese dieses Abschnittes und die anschließende Ligation.
NER kommt sowohl bei Pro- als auch bei Eukaryoten vor, jedoch unterscheiden sich die Mechanismen und beteiligten Enzyme. Während bei Prokaryonten wie Escherichia coli Uvr Proteine und DNA-Polymerase I beteiligt sind, sind es bei Eukaryonten Proteine, die ihre Namen von den Erbkrankheiten Xeroderma pigmentosum und Cockayne-Syndrom haben, z. B. XPA bzw. CSA. Zu den beteiligten Polymerasen gehören die DNA-Polymerasen δ, ε und/oder κ.
Bei Eukaryonten gibt es zwei Wege der Nukleotidexzisionsreparatur. Zum einen Global Genome Repair (GGR), welche Schäden in transkriptionsinaktiven Bereichen der DNA behebt und zum anderen die sogenannte Transcription Coupled Repair (TCR), welche Schäden an der aktuell zu transkribierenden DNA behebt. Diese beiden Formen unterscheiden sich nur in der Schadenserkennung.
Bei der TCR ist es von Bedeutung, dass die durch die Schädigung blockierte RNA-Polymerase II entfernt wird, um so den TCR-Proteinen Zugriff zur DNA-Schädigung zu ermöglichen. Dieses Entfernen der RNA-Polymerase II wird durch CSA und CSB ermöglicht. Bei der GGR wird die DNA-Läsion vom Proteinkomplex XPC/HHR23B erkannt. Dagegen spielt dieser Komplex bei der TCR keine Rolle. Die weiteren Schritte sind bei beiden Reparaturwegen identisch. Zur weiteren DNA-Schadenserkennung dienen XPA und RPA und sie dirigieren die Helikasen XPB und XPD zur Läsion, welche unmittelbar in der Nähe der Schädigung die DNA entwinden.
In den letzten Jahren wurde durch die Medien vermehrt auf das Schicksal der Mondscheinkinder hingewiesen. Es handelt sich dabei um Kinder, die von einem seltenen genetischen Defekt betroffen sind und, um der schnellen Entstehung von Hautkrebs entgegenzuwirken, jegliche Exposition von Sonnenlicht vermeiden müssen. Diese Kinder leiden unter Xeroderma Pigmentosum (XP, auch Melanosis lenticularis progressiva oder Mondscheinkrankheit genannt), das durch einen Defekt in der Nukleotidexzsionsreparatur entsteht. Durch den Defekt kommt es aber nicht nur zu Xeroderma Pigmentosum, sondern es entstehen zwei weitere Krankheiten, das sogenannte Cockayne-Syndrom und Trichothiodystrophie.
Die Endonukleasen XPG und XPF-ERCC1 schneiden den DNA-Strang an zwei Stellen, 3' bzw. 5' (duale Inzision), so dass ein ca. 30 Basen umfassendes Oligonukleotid freigesetzt wird, welches die Schädigung enthält. Nun folgt die Polymerisation des fehlenden DNA-Abschnitts durch DNA-Polymerase und weitere Faktoren. Als letztes erfolgt die Ligation des synthetisierten Abschnitts durch DNA-Ligase I und Flap Endonuklease 1 oder den Ligase-III-XRCC1-Komplex.
Mutationen betreffend der XPA-XPG Familie führen zur Ausbildung des Krankheitsbildes Xeroderma pigmentosum. Bei Xeroderma pigmentosum ist das Hautkrebsrisiko erhöht, was auf die Wichtigkeit einer funktionierenden DNA-Reparatur nach UV-Bestrahlung hinweist.
Korrekturlesen durch DNA-Polymerase (Basenfehlpaarungsreparatur)
Bei der Replikation werden die beiden Stränge eines DNA-Doppelstrangs getrennt und jeweils durch Basenpaarung komplementär ergänzt, sodass anschließend zwei (nahezu) identische doppelsträngige DNA-Moleküle vorliegen. Das hieran beteiligte Enzym, eine DNA-abhängige DNA-Polymerase, katalysiert nicht nur die Synthese eines neuen DNA-Strangs aus Nukleotiden anhand der vorliegenden DNA-Matrize. Es kann zudem während des Prozesses eine Fehlpaarung (englisch mismatch) erkennen und rückgängig machen, sodass das korrekt gepaarte Desoxyribonukleotid angefügt werden kann; dies wird als Korrekturlesen (englisch proofreading) bezeichnet.
Diese zusätzliche Funktion der DNA-Polymerase wird mit sehr geringer Fehlerquote ausgeführt, allerdings nicht hundertprozentig genau. Durch die Mithilfe von DNA-Mismatch-Reparaturproteinen wird die Fehlerquote weiter abgesenkt, die Anzahl spontaner Mutationen liegt damit um rund das Tausendfache niedriger. Anhand seines Methylierungsstatus kann hierbei ein womöglich fehlerhaft entstandener Tochterstrang von der Elternstrangmatrize unterschieden werden, da er erst später methyliert wird. Ein Defekt im Ablauf der Mismatchreparatur kann beispielsweise eine Form von Darmkrebs, ein hereditäres non-polypöses kolorektales Karzinom, verursachen.
Photoreaktivierung
Photolyasen sind in der Lage, durch ultraviolette Strahlung in der DNA entstandene Cyclobutan-Ringe und (6-4)-Photoprodukte aufzulösen. Sie verfügen über einen sog. Antennenkomplex, mit dem sie blaues oder ultraviolettes Licht absorbieren und mithilfe dieser Energie vom Kofaktor FAD zwei Elektronen auf den im aktiven Zentrum des Enzyms gebundenen DNA-Schaden übertragen. Selbiger spaltet sich in der Folge. Die Photolyase stellt so ohne Herausschneiden und Einfügen von Basen die native Struktur der DNA wieder her. Bis heute konnten in vielen Organismen von den Prokaryoten über Pilze und Pflanzen bis hin zu Beuteltieren Photolyasen nachgewiesen werden. Trotz ihrer unbestrittenen vorteilhaften Eigenschaften für diese Organismen sind die Photolyasen im Laufe der Evolution mehrfach verloren gegangen.[6] Auch die höheren Säugetiere, zu denen der Mensch zählt, besitzen keine reparaturaktiven Varianten dieser Proteine mehr. Die Gründe hierfür sind bisher nicht abschließend geklärt.
Reparatur von Doppelstrangbrüchen
Die Reparatur eines Doppelstrangbruchs kann generell über homologe Reparaturmechanismen erfolgen oder durch nicht-homologe Reparatur. Nicht-homologe Reparatur ist dabei fehleranfälliger und führt häufiger zu Veränderungen der Ursprungssequenz in Form von Deletionen oder kleineren Insertionen. Diese Eigenschaft macht man sich bei der Genom-Editierung bspw. durch das CRISPR/Cas9-System zunutze um zielgerichtet Mutationen in einem Genom zu erzeugen. Homologe Reparaturmechanismen sind insbesondere in Bakterien und Hefen verbreitet, während in höheren Eukaryoten die meisten Doppelstrangbrüche nicht-homolog repariert werden. Dabei sind homologe Reparaturmechanismen besonders während der S und G2-Zellphase aktiv. Das Schwester-Chromatid ist dann häufig nahe der Bruchstelle und kann als Template zur Reparatur dienen. In der G1 bis frühen S-Phase ist nicht-homologe Reparatur aktiver.
Homologe Reparatur
Homologe Reparatur beginnt mit der Resektion der 5'-Enden eines offenen Bruches durch den MRX-Komplex (MRN in Säugern und Pflanzen) bestehend aus MRE11, Rad50 und XRCC1 (NBS1). U. a. mit Hilfe der Proteine Rad51, Rad54 bindet das einzelsträngige 3'-Ende des Bruchs in einem zur Reparaturstelle homologen Bereich (bspw. dem Schwester-Chromatid) und bildet eine D-Loop genannte Struktur aus. Anhand der homologen Vorlage wird der fehlende Strang synthetisiert. Daraufhin bilden sich zwei Holliday-Strukturen aus, deren Auflösung entweder ein Crossover der beiden Stränge oder eine Auflösung zur Folge hat.
Synthesis dependant strand-annealing (SDSA)
Findet die Bindung des offenen Endes des Doppelstrangbruchs in einem nicht zur Reparaturstelle homologen Bereich statt, spricht man von SDSA. Dies hat zur Folge, dass der Bruch zwar repariert wird, aber sogenannte Filler-Sequenzen eingebaut werden, die aus dem Bereich stammen, an dem sich der D-Loop gebildet hat.
Single strand annealing
Bei Bruch innerhalb zwischen Repeats kann es dazu kommen, dass die Enden des Bruches soweit verkürzt werden, dass homologe Paarung an längeren Bereichen von Homologie erfolgen kann. Der Strang wird nachfolgend repariert, was zu einer Deletion des Bereiches zwischen den beiden Repeats führt.
Nicht-homologe Reparatur
Nicht-homologe Reparatur erfolgt ohne Verwendung einer homologen Vorlage. Zu unterscheiden sind Non-homologous end-joining (NHEJ) und Microhomology-mediated end-joining (MMEJ) – manchmal auch alternative- oder backup-NHEJ genannt.
Non-homologous end-joining (NHEJ)
NHEJ startet durch Bindung des Ku-Komplexes bestehend aus Ku70 und Ku80 an die offenen Enden des Doppelstrangbruchs. Dadurch sind die offenen Enden vor weiterem Abbau durch Exonukleasen geschützt und der Bruch wird stabilisiert. Bei der Bindung der offenen Enden werden oft kurze Bereiche von Homologie zwischen den offenen Enden (sogenannte Mikrohomologie) ausgenutzt. Anschließend erfolgt die Bindung des MRX bzw. MRN-Komplexes (siehe homologe Reparatur), der die offenen Enden prozessiert, sodass sie durch den XRCC4-DNA-LigaseIV-Komplex repariert werden kann. NHEJ wird oft als fehleranfälliger Reparaturmechanismus beschrieben. Die Folge einer Reparatur durch NHEJ sind meistens kleinere Deletionen von wenigen Basen oder kleine Insertionen an der Reparaturstelle.
Microhomology-mediated end-joining (MMEJ)
Bindet der Ku-Komplex nicht an ein offenes Ende, bspw. in Knockout-Mutanten oder während der S oder G2-Phase, wenn Ku70/80 nicht so aktiv ist, findet die Stabilisierung der offenen Enden an längeren Bereichen von Mikrohomologie statt (5-25 bp). Anschließend wird der offene Bruch durch den MRX/MRN-Komplex prozessiert und durch XRCC4/DNA-LigaseIV repariert. Ohne den Schutz durch den Ku-Komplex, sind die offenen Enden länger Exonukleasen ausgesetzt. Dadurch und durch die Verwendung von größeren Bereich von Mikrohomologie an der Reparaturstelle können auch größere Deletionen entstehen.
Eine Störung dieser Reparatursysteme manifestiert sich klinisch häufig als Chromosomenbruchsyndrom, wie zum Beispiel das Nijmegen-Breakage-Syndrom.
Reparatur von Quervernetzungen
Quervernetzte DNA führt in Wirbeltierzellen zu einer Aktivierung der Proteine RAD18-SLF1-SLF2, wodurch eine Ubiquitinierung der vernetzten DNA durch RNF8/RNF168 erfolgt und anschließend der SMC5/6-Proteinkomplex bindet.[7]
Einzelnachweise
- C. R. Bartram: Genetische Grundlagen der Kanzerogenese. In: W. Hiddemann, C. R. Bartram (Hrsg.): Die Onkologie. Teil 1, Ausgabe 2, Verlag Springer, 2009, ISBN 3-540-79724-6, S. 118–127 (eingeschränkte Vorschau in der Google-Buchsuche).
- S. Uphoff, R. Reyes-Lamothe u. a.: Single-molecule DNA repair in live bacteria. In: Proceedings of the National Academy of Sciences. Band 110, Nummer 20, Mai 2013, S. 8063–8068, doi:10.1073/pnas.1301804110. PMID 23630273. PMC 3657774 (freier Volltext).
- Sung-Lim Yu, Sung-Keun Lee: Ultraviolet radiation: DNA damage, repair, and human disorders. In: Molecular & Cellular Toxicology. 13, 2017, S. 21, doi:10.1007/s13273-017-0002-0.
- Peter Elsner, Erhard Hoelzle u. a.: Täglicher Lichtschutz in der Prävention chronischer UV-Schäden der Haut. In: JDDG. 5, 2007, doi:10.1111/j.1610-0387.2007.06099_supp.x.
- Rolf Sauer: Strahlentherapie und Onkologie. 5. Auflage, Elsevier GmbH, Urban und Fischer Verlag, München, 2010; ISBN 978-3-437-47501-6, S. 112 (eingeschränkte Vorschau in der Google-Buchsuche).
- Lucas-Lledó, J.I. & Lynch, M. Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family. Mol. Biol. Evol 26, 1143–1153 (2009).
- M. Raschle, G. Smeenk, R. K. Hansen, T. Temu, Y. Oka, M. Y. Hein, N. Nagaraj, D. T. Long, J. C. Walter, K. Hofmann, Z. Storchova, J. Cox, S. Bekker-Jensen, N. Mailand, M. Mann: Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. In: Science. 348, 2015, S. 1253671, doi:10.1126/science.1253671.