Zeolith Y
Zeolith Y ist eine technisch bedeutsame, künstlich hergestellte kristalline Substanz aus der Stoffgruppe der Zeolithe. Unter den Mineralen hat es seine Entsprechung im Faujasit. Die wichtigste Anwendung von Zeolith Y ist als Katalysator im Fluid Catalytic Cracking (FCC-Verfahren) in der Raffinerie zur Herstellung leichtsiedender Fraktionen (Benzin und Diesel) aus Rückständen der Erdöldestillation.
Geschichte
Nach der Synthese von Zeolith A und Zeolith X mit einem Si/Al-Verhältnis nahe 1 im Jahr 1950 durch Robert Milton bei Linde Air Products Division von Union Carbide wurde bemerkt, dass synthetische Zeolithe anfällig gegen Säuren, Wasser oder Dampf aufgrund von Dealuminierung waren.[1] Mordenit, mit einem Si/Al-Verhältnis von 5, ist weniger anfällig. Während der Bemühungen, synthetische Zeolithe mit einem höheren Si/Al-Verhältnis herzustellen, gelang es 1954 Donald Breck, einem Kollegen von Milton bei Union Carbide, Zeolith Y zu synthetisieren. Breck erhielt 1964 ein Patent zur Herstellung von kristallinem Zeolith Y.[2] Zeolith Y zeigte, wie erwartet, eine höhere Stabilität. Uytterhoeven et al. bestimmten 1965 als erste den Protonengehalt von HY, der sauren From von Zeolith Y.[3][4] 1967 zeigten George Kerr und seine Mitarbeiter von Mobil und W. R. Grace and Company, dass eine ultrastabile Form von Zeolith Y (USY) durch Erhitzen in einer „inerten statischen Atmosphäre“ hergestellt werden konnte.[5] Im selben Jahr veröffentlichte Benesi die ersten thermogravimetrischen Kurven zur thermischen Zersetzung der Ammoniumform von Zeolith Y.[6][4]
Aufbau
Kristallstruktur
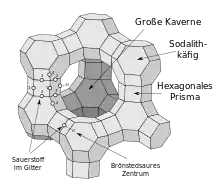

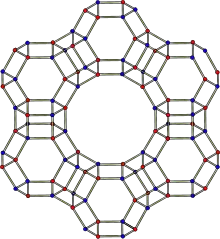
Die Kristallstruktur des Zeolith Y entspricht derjenigen von Faujasit. Das Faujasitgerüst besteht aus Sodalithkäfigen, die über hexagonale Prismen miteinander verbunden sind. Die Poren, die durch den Verbund dieser Sekundärstrukturen gebildet werden, sind senkrecht zueinander angeordnet. Die größte Pore, die durch einen Ring mit 12 Einheiten gebildet wird, hat einen Durchmesser von 7,4 Å und ist damit im Vergleich zu anderen Zeolithen relativ groß. Der Innenraum der großen Kaverne hat einen Durchmesser von 12 Å und ist umgeben von 10 Sodalithkäfigen. Die Zelleinheit ist kubisch mit einer Länge von 24,7 Å,[8] wobei diese Länge abhängig vom Si/Al-Verhältnis, Art und Konzentration der Gegenkationen und Hydratiserungsgrad zwischen 24,2 Å und 25,1 Å schwanken kann.[9] Die Pore, die durch das hexagonale Prisma gebildet wird, hat einen Durchmesser von 2,5 Å und ist damit nur etwa ein Drittel so groß wie die große Pore.[10]
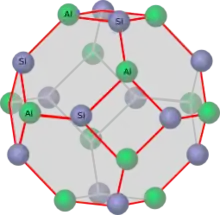
Sodalithkäfige und hexagonale Prismen (Sekundärstrukturen) bilden Elementarzellen (Tertiärstrukturen). Eine Elementarzelle enthält 18 Sodalithkäfige und 16 hexagonale Prismen. Eine solche Elementarzelle hat die Zusammensetzung Na58Al58Si134O125* 240 H2O.
Zeolith Y der Na-Form enthält 52 Natriumionen pro Elementarzelle, davon 16 innerhalb der hexagonalen Prismen und der Sodalithkäfigen.[11] Die Natriumionen in den hexagonalen Prismen sind mit 6 Sauerstoffionen aus dem Zeolithgerüst koordiniert. In diesen Prismen gibt es keinen weiteren Platz für Wassermoleküle, so dass die Natriumionen mit den Sauerstoffionen des Zeolithgerüstes koordinieren. In den Sodalithkäfigen sind sie mit 3 Sauerstoffionen aus dem Zeolithgerüst und zwei Wassermolekülen koordiniert. Die restlichen 36 Natriumionen befinden sich in den Superkäfigen und sind mit 6,5 Molekülen Wasser vollständig hydratisiert. Diese Natriumionen sind mobil und verhalten sich wie in einer wässrigen Lösung.
Das Si/Al-Verhältnis beträgt 2,43 und der Leervolumenanteil 48 %.[12]
Faujasit-Materialien zeichnen sich durch eine große Oberfläche und eine enge Porenverteilung im Bereich von 0,9 bis 1,2 nm, sowie durch eine hohe thermische Beständigkeit aus.[13] Zeolith Y zersetzt sich ab 793 °C.[8]
Aluminiumgehalt und Aluminiumverteilung
Das Gerüst von Zeolithen besteht aus Silizium und Aluminiumatomen, die über Sauerstoffatome miteinander verbunden sind. Bei „normalem“ Zeolith Y beträgt das Si/Al-Verhältnis ca. 2,5. Allerdings ist es möglich, Aluminiumatome durch Siliziumatome zu ersetzen. Dies kann durch eine gezielte Dealuminierung erfolgen. Nach Dealuminierung unterscheidet man zwischen Gitteraluminium und Aluminium außerhalb des Gitters. Das Aluminium außerhalb des Gitters bleibt in den meisten Fällen in den Hohlräumen des Zeolithes erhalten.[14] Durch die Dealuminierung schrumpft die Elementarzelle und der Zeolith wird stabiler.
Während zwei Siliziumatome über einen Sauerstoff verbunden nebeneinander sein dürfen, muss neben jedem Aluminiumatom ein Siliziumatom sein. Zwei benachbarte Aluminiumatome sind nicht erlaubt. Bei dehydratisierten Zeolithen der H-Form bildet die Hydroxygruppe zwischen Silizium und Aluminium das brönstedsaure Zentrum:
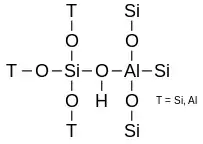
Direkt nach der Synthese steht an dieser Stelle ein Natriumion als Gegenkation, nach dem Ionentausch das entsprechende Ion, meistens Ammonium oder Lanthan.
Saure Zentren
Jedes koordinierte Aluminiumatom innerhalb des Gittergerüstes hat eine negative Ladung mit einem Gegenkation an bestimmten Positionen außerhalb des Gittergerüstes. In der dehydratisierten Form bestehen die Gegenkationen aus Protonen (Brönstedsaure Zentren) an bevorzugten Positionen, die zwei unterschiedliche saure Hydroxygruppen bilden:
- Hydroxygruppen im Superkäfig. Sie sind stark sauer und direkt zugänglich zu Adsorbaten.
- Hydroxygruppen in den Sodalithkäfigen. Sie sind weniger sauer, aber mobil genug, um mit Adsorbaten in den Superkäfigen in Wechselwirkung zu treten.[15]
Die brönstedsauren Zentren haben nicht alle die gleiche Stärke.[7] Diese hängt vom Aluminiumgehalt ab. Bei einem Si/Al-Verhältnis größer als 4,5 haben alle Zentren die gleiche Stärke. Bei einem höheren Aluminiumgehalt (niedrigerer Si/Al-Wert als 4,5) gibt es Zentren schwacher und mittlerer Aktivität. Dies macht sich in den katalytischen Eigenschaften des Zeolithes bemerkbar. Befinden sich zwei Aluminiumatome in unmittelbarer Nachbarschaft eines Siliziumatomes, wird die Stärke der brönstedsauren Zentren reduziert. Aus diesem Grund nimmt die Stärke der einzelnen brönstedsauren Zentren mit zunehmendem Aluminiumgehalt ab.
Der Aluminiumgehalt wirkt sich außerdem auf die Schwingfrequenz der O-H Dehnung der brönstedsauren Zentren aus, wie das IR-Spektrum zeigt. Diese unterschiedlichen Zentren werden unterschiedlichen kristallographischen Positionen zugeschrieben. Im ersten Fall zeigt der Proton in Richtung große Kaverne, im zweiten Fall befindet er sich innerhalb des Sodalithkäfiges (siehe Bild mit Positionen der Sauerstoffatomen).
Formen
Je nach Gegenion am Aluminium wird nach „Formen“ unterschieden:
- NaY ist die ursprüngliche Form mit Natrium.
- HY ist die natriumfreie, in den sauren Zustand überführte Form. Diese Bezeichnung wurde 1969 von Kerr et al. eingeführt.[16][4]
- NH4Y ist die Form mit Ammonium als Gegenion. Durch Kalzinierung entsteht daraus HY.
Weiterhin unterscheidet man nach der Stabilität:
- USY für ultrastabiler Zeolith Y (ultra stable), der durch Behandlung mit Dampf bei hohen Temperaturen gewonnen wird, indem eine Dealuminierung des Gitters stattfindet.[17]
- VUSY für very ultrastable
Matrix
Die Zeolithkristalle sind zu klein, als dass sie als solche in der FCC-Unit direkt eingesetzt werden könnten. Um sie aus dem Reaktionsgemisch abscheiden zu können, benötigt man Partikel mit einer Mindestgröße. Die Zeolithkristalle werden daher in eine Matrix eingebettet. Diese Matrix erhält die Form einer Mikrokugel, sog. „microspheres“ mit einer Größe von ca. 70 µm. Diese können im Abscheider der FCC-Unit durch Schwerkraft abgetrennt werden.
Die Matrix kann katalytisch inert oder aktiv sein.
Katalytisch inerte Matrizen
Unter den katalytisch inerten Matrizen unterscheidet man zwei Typen:[15]
- Aus Kieselgel abgeleitet
- Aus Tonerde abgeleitet
Kieselgel-abgeleitete Matrizen
Ein Silica hydrosol (eine kolloidale Suspension) wird durch Ansäuerung auf pH 3 einer Natriumsilikatlösung hergestellt. Die Zeolithkristalle und feine Tonerde werden eingerührt. Das Gemisch wird dann sprühgetrocknet und die dadurch entstandenen Mikrokugeln durch Ionentausch und Kalzinierung weiterverarbeitet. Das Endprodukt enthält den aktiven Zeolithen in einer Matrix aus amorphem Kieselgel und Tonerde. Ein solcher Katalysator ist kostengünstig und abriebfest, aber die Matrix hat nur kleine Poren und eine geringe innere Oberfläche, die keinen nennenswerten Beitrag zur Crack-Reaktion leistet.
Tonerde-abgeleitete Matrizen
Eine solche Matrix wird durch Kristallisation in situ, d. h. innerhalb der Matrix selbst, erzeugt. Diese Technologie wurde von der Engelhard Corporation ausgearbeitet.[18][19][20] Hierzu wird gemahlenes, hydratisiertes Kaolin bei ca. 1000 °C kalziniert, wodurch es zum Spinell umgewandelt wird. Das kalzinierte Material wird mit weiterem hydratisierten Kaolin, einer geringen Menge Natriumsilikat und Zeolithkeime vermischt. Das resultierende Gemisch wird sprühgetrocknet und die sich daraus ergebenden Mikrokugeln bei 732 °C kalziniert, um das hydratisierte Kaolin in Metakaolin (siehe auch Kaolinit) umzuwandeln. Das Spinell und das Metakaolin werden innerhalb der Mikrokugel durch das Natriumsilikat als Bindemittel zusammengehalten. Die kalzinierten Mikrokugeln werden anschließend in einer Mischung aus Natriumsilikat und Natronlauge behandelt, wobei ein Teil der Matrix (das Metakaolin) zum Zeolith Y kristallisiert.[15]
Katalytisch aktive Matrizen
Unter den katalytisch aktiven Matrizen unterscheidet man zwei Typen:[15]
- Aluminiumoxid-sol-abgeleitete Matrizen
- Matrizen aus amorphem Siliziumdioxid/Aluminiumoxid
Aluminiumoxid-sol-abgeleitete Matrizen
Die katalytisch aktive Matrix besteht aus amorphem Aluminiumoxid. Der Zeolith wird vor der Zugabe in die Matrix ionengetauscht und modifiziert. Pseudobömit wird dabei zunächst mit einer Säure peptisiert und anschließend mit Ton und dem geeigneten Y-Zeolith vermengt. Das sprühgetrocknete Produkt hat eine katalytisch aktive Matrix mit hoher spezifischer Oberfläche, die die Oktanzahl vom Produkt erhöht und beim Cracking von Rückstand effektiv ist.
Matrizen aus amorphem SiO2/Al2O3
Diese Matrizen können aus einem SiO2/Al2O3-Hydrogel oder -Hydrosol hergestellt werden. Eine solche Matrix erhöht die katalytische Aktivität und die Oktanzahl.
Synthese
Industriell wird Zeolith Y durch Kristallisation hergestellt, wobei im Allgemeinen ein Ionentausch mit anschließender Kalzinierung bei milder Temperatur und ein zweiter Ionentausch mit einer zweiten Kalzinierung bei höherer Temperatur folgen. Es gibt mehrere Herstellverfahren, die sich aber nach dem folgenden Schema verallgemeinern lassen.
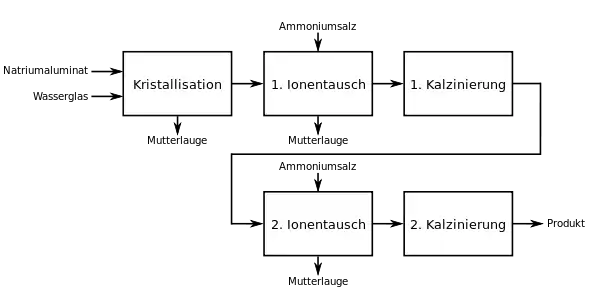
Für die Kristallisation setzt man eine Aluminium- (Natriumaluminat) und eine Siliziumquelle (Natriumsilikat, Wasserglas) ein. Es können aber auch andere Stoffe eingesetzt werden, die beide Elemente enthalten, z. B. Kaolin. In der Natur vorkommendes Kaolinit ist aufgrund der niedrigen Kosten die bevorzugte Quelle. Dieses ist jedoch als solches nicht einsetzbar, sondern muss vorher bei hoher Temperatur (700 °C bis 900 °C) kalziniert werden, um es durch Umwandlung zu Metakaolin chemisch aktiv zu machen.[21]
Im ersten Ionentausch wird der Natriumgehalt von ca. 9 % auf ca. 2 % verringert. Durch die erste Kalzinierung werden die verbleibenden Natriumionen „mobilisiert“ und für einen zweiten Ionentausch zugänglich gemacht. In der zweiten Kalzinierung werden die Ammoniumionen als Ammoniak entfernt, wobei saure Zentren zurückbleiben.
Kristallisation
Zeolith Y wird – wie auch andere Zeolithe – durch Kristallisation aus Aluminium (z. B. Natriumaluminate) und Silicium enthaltenden Stoffen (z. B. Natriumsilikate oder Wasserglas) hergestellt. Auch andere Alumosilikate wie Kaolin können als Aluminium- und Siliciumquellen dienen. Die Edukte werden in einem alkalischen Medium, z. B. Natronlauge, bei Temperaturen zwischen 70 °C und 300 °C (meistens um 100 °C) gelöst und kristallisiert. Dabei handelt es sich nicht um eine Kristallisation im klassischen Sinne, bei der die Löslichkeitsgrenze eines gelösten Stoffes überschritten wird, so dass dieser in den festen Zustand übergeht, sondern vielmehr um eine chemische Reaktion, bei der die einzelnen Komponenten (Aluminate und Silikate) in Form eines Kristallgitters zusammengefügt werden.
Ionentausch
Nach der Synthese liegt der Zeolith in seiner Natriumform vor, d. h. die Gegenionen der Aluminiumionen bestehen aus Natriumkationen. Er wird durch Ionentausch mit wässrigen Lösungen aus Ammoniumsalzen (z. B. Ammoniumnitrat oder Ammoniumsulfat) in die Ammoniumform überführt. Die Natriumkationen werden durch Ammoniumkationen ersetzt und gelangen in die Mutterlauge. Der Ionentausch kann beispielsweise durch Rühren in Behältern unter Zugabe der Ammoniumsalzlösung durchgeführt werden.

Die Natriumkationen in den Superkäfigen können leicht ausgetauscht werden, da sie hydratisiert und mobil sind. Die Natriumionen in den hexagonalen Prismen sind dagegen mit den Sauerstoffionen des Zeolithgerüstes koordiniert und daher nur schwer zugänglich. Sie lassen sich nicht ohne weiteres ionenaustauschen. Diese Natriumionen werden durch eine Kalzinierung bei relativ milder Temperatur (600 °C)[23] „mobilisiert“ und können in einem anschließenden, zweiten Ionentausch ausgetauscht werden. Die Natriumionen migrieren während der Kalzinierung außerhalb der hexagonalen Prismen. Dabei wandert Aluminium als Gegenion für das Natrium mit, wodurch es zu einer Dealuminierung des Gerüstes kommt, bei der Mesoporen entstehen. Durch den zweiten Ionentausch kann der Natriumgehalt auf Größenordnungen um 1000 ppm reduziert werden, was für die Stabilität des Endproduktes wichtig ist. Philip Maher und Carl McDaniel von W. R. Grace and Company erhielten 1975 ein Patent zum verbesserten Ionentausch an kristallinen Zeolithen durch Zwischenkalzinierung.[22]
Der Ionentausch wird außerdem durch den pH-Wert beeinflusst.[23] Der Austauschgrad von Natrium ist höher, je tiefer (saurer) der pH-Wert ist. Problematisch dabei ist die Zerstörung der Kristallstruktur bei zu niedrigen pH-Werten. So wurde gefunden, dass beim ersten Ionentausch kein Kristallinitätsverlust zwischen pH 3,0 und 4,8 entsteht. Es ist daher vorteilhaft, den ersten Ionentausch bei einem pH-Wert von 3,0 durchzuführen. Im zweiten Ionentausch wurde dagegen bei einem pH-Wert von 3,0 bereits eine Abnahme der Kristallinität auf 70 % gefunden. Der zweite Ionentausch sollte daher bei einem höheren pH-Wert von 4,5 durchgeführt werden.
Um die Zeolithstruktur zu stabilisieren, wird in der Regel ein Teil des Natriums durch Seltenerdmetalle, meistens Lanthan, teilweise auch Cer, ebenfalls durch Ionentausch ersetzt. Der Ersatz des einwertigen Natriumions durch zwei- oder dreiwertige Kationen führt zu einer Erhöhung der Gitterenergie und erhöht dadurch die Strukturstabilität.[24] Bei 25 °C sind die hydratisierten Lanthanionen zu groß, um von den Superkäfigen in die hexagonalen Prismen diffundieren zu können.[25] Die hexagonalen Prismen haben einen freien Durchmesser von 0,25 nm, während die hydratisierten La3+-Ionen mit 0.396 nm (0,79 nm laut[26]) größer sind. Damit Lanthan in die kleinen Sodalithkäfigen durch die hexagonalen Prismen diffundieren kann, muss die Hydrathülle durch Erhöhung der Temperatur auf 82 °C mobilisiert werden[27] (50 °C bis 90 °C laut[26]).
Ein Festphasen-Ionentausch ist ebenfalls möglich, aber eher eine Seltenheit und ohne technische Bedeutung. Hierzu wird das trockene Salz mit dem Zeolith bei hoher Temperatur in Kontakt gebracht. Diese Methode kann angewandt werden, um Zeolithe der H-Form mit Lanthan auszutauschen, z. B. mit LaCl3. Die Salzmoleküle diffundieren dabei zu den Austauschstellen.[28]
Kalzinierung
Durch anschließende Kalzinierung wird das Ammoniumion entfernt und der Zeolith in seine saure Form überführt. In der sogenannten Ultrastabilisierung wird der Zeolith bei 700 °C bis 800 °C mit Wasserdampf behandelt.[12] Dabei verliert der Zeolith Aluminium und die spezifische Oberfläche geht zurück.
Durch die Kalzinierung verlieren die Gegenkationen, speziell Natrium oder Lanthan, ihre Hydrathüllen. Dadurch können sie durch die hexagonalen Prismen in die kleineren Sodalithkäfige diffundieren. So können beispielsweise Lanthanionen ab Temperaturen um 100 °C in die Sodalithkäfige diffundieren.[25] Bei Temperaturen von höher als 200 °C in Gegenwart von Lanthan, bildet sich an jedem Lanthanion eine Hydroxygruppe und ein Proton. Die dadurch gebildete Säure führt zu einer Dealuminierung des Gitters.
Dealuminierung
Das atomare Verhältnis Si/Al in Zeolithen stellt einen wichtigen Parameter dar, der solche Eigenschaften wie maximale Ionentauschfähigkeit, thermische und hydrothermale Stabilität, Hydrophobizität, sowie Anzahl und Stärke der brönstedsauren Zentren definiert. Zeolithe mit einem niedrigen Aluminiumgehalt sind in der Regel stabiler, vor allem wenn sie als FCC-Katalysatoren verwendet werden. Da die Synthese von Zeolith Y nur in einem engen Si/Al-Bereich ohne größeren Aufwand gelingt, kann dieser nicht mit einem Si/Al-Verhältnis größer als 2.5 direkt durch Kristallisation hergestellt werden.[29] Das Si/Al-Verhältnis kann jedoch durch spätere Entfernung von Aluminium aus dem Gitter erhöht werden. Dies führt zu Gitterfehlstellen, die teilweise durch Einbau anderer Elemente, insbesondere Silizium, erneut besetzt werden können. Durch die Dealuminierung nimmt die Größe der Zelleinheit leicht ab, da die SiO-Bindungen kürzer als die Al-O-Bindungen sind.[30]
Es gibt verschiedene Dealuminierungsverfahren, wovon die wichtigsten die folgenden sind:
- Hydrothermale Behandlung (mit heißem Wasserdampf)
- Säurebehandlung
- Behandlung mit gasförmigen Halliden oder Halogenen
- Komplexierung mit chelatisierenden Agentien wie EDTA
Durch den Wegfall der AlO2-Gruppen verringert sich die Anzahl an (Natrium-)Gegenkationen. Dies verringert auch die maximal mögliche Beladung mit Metallen der seltenen Erden.

Die Dealuminierung führt auch zur Bildung von Mesoporen, wodurch die katalytischen Eigenschaften während des Crackens verbessert werden.[23]
Die Aluminiumatome, die aus dem Zeolithgitter entfernt werden, bilden außerhalb des Gitters Aluminiumspezies, die mit der Bildung oktaedrisch koordiniertem Aluminiums in Verbindung gebracht wurden.[31] Es wurde vorgeschlagen, dass diese Aluminiumspezies eine separate Phase außerhalb des Zeolithkristalls bilden. Alternativ wurde vorgeschlagen, dass sie als isolierte Kationen innerhalb der Zeolithkäfige vorhanden sind und die elektrische Ladung kompensieren. Außerhalb des Gitters kann sich auch tetraedrisch koordiniertes Aluminium befinden. Die Gegenwart von Aluminiumspezies außerhalb des Gitters führt zu einer besseren hydrothermalen Stabilität, erhöhter Lewisazidität sowie erhöhter katalytischer Aktivität bei Reaktionen, die starke Brönsted Säuren benötigen, wie z. B. das Cracken.
Hydrothermale Dealuminierung
Die hydrothermale Behandlung ist die technisch bevorzugte Variante, um Aluminium aus dem Gerüst zu entfernen und die Stabilität des Zeolithen zu erhöhen. Bei der Dealuminierung durch Kalzinierung mit Wasserdampf bei hohen Temperaturen wandert ein Teil des Aluminiums im Gitter zu Stellen außerhalb des Gitters:

Es handelt sich im Wesentlichen um eine Hochtemperaturhydrolyse der Si-O-Al-Bindungen unter Bildung von Aluminium-Spezies außerhalb des Gittergerüstes.[32][15] Das gebildete Aluminiumhydroxid kann als amphoterer Stoff mit den sauren Zentren auf dem Zeolith reagieren und ist durch Ionentausch zugänglich:

Unter diesen Bedingungen findet eine Umstrukturierung des Zeolithgerüstes unter Beseitigung der entstandenen Fehlstellen statt. Diese werden durch nachrückendes Silizium besetzt:
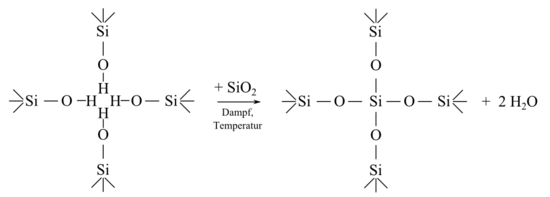
Das Ergebnis ist ein ultrastabiler Y-Zeolith (USY) mit höherem Siliziumgehalt, der sich durch eine hohe thermische und hydrothermale Stabilität, eine kleinere Zelleinheit und ein verringertes Ionenaustauschvermögen auszeichnet.[17] Für diese Art der Dealuminierung verwendet man in der Regel natriumfreien Zeolith der Ammonium-Form. Die Kalzinierung wird bei Temperaturen zwischen 500 °C und 870 °C durchgeführt.
Desilifizierung
Unter Desilifizierung versteht man die Entnahme von Siliziumatomen aus dem Zeolithgitter. Nach der intensiven Untersuchung der Dealuminierung von Zeolithen, nimmt das Interesse an der Desilifizierung zu. Hierdurch kann post-synthetisch die Mesoporosität erhöht werden.[33] Dies verbessert die Diffusionseigenschaften des Zeolithes. Im Allgemeinen werden Zeolithe durch Behandlung mit Natronlauge desilifiziert.
Eigenschaften
Beweglichkeit der Ionen
In der hydratisierten Form sind die Ionen und Wassermolekülen im Superkäfig sehr beweglich, was sowohl einen Ionentausch als auch eine reversible Dehydratisierung und Sorption erlaubt.
Stabilität
Zeolith Y der H-Form, der durch Kalzinierung der Ammoniumform gewonnen wird, ist in Gegenwart von Wasser sehr instabil.[29] Diese Instabilität wurde auf die intrinsische Acidität dieser Form zurückgeführt, wenn Gitter-Wasserstoffionen mit interkristallinem, „flüssigem“ Wasser H3O+-Ionen bilden. Dies führt zu einer Dealuminierung des Gitters ähnlich der, die durch eine Behandlung mit Säure entsteht.
Die thermische Stabilität des Zeolithgitters hängt wesentlich von dem Gegenkation am Aluminium, dessen Verteilung im Gitter und dem Grad an Ionentausch ab.[24] Ionentausch mit multivalenten Kationen, insbesondere die der Metalle der seltenen Erden, erhöhen die thermische Stabilität von Zeolith Y (siehe Synthese und Ionentausch).
Ultrastabiler Zeolith (USY)
Um als Katalysator im FCC-Verfahren geeignet zu sein, muss das Material bei Temperaturen um 700 bis 950 °C in Gegenwart von Wasserdampf beständig sein.[30] Obwohl die Bereitstellung von natriumfreiem Y-Zeolith (HY) einen wesentlichen Fortschritt darstellte, war er nur bis Temperaturen um 500 °C beständig. Es wurde bemerkt, dass in Gegenwart von Luftfeuchte sogar bei Raumtemperatur die Struktur von HY beschädigt wurde.[30][34] Die Stabilität konnte durch Ionentausch mit mehrwertigen Kationen erhöht werden.[30][35] Parallel dazu wurden andere Methoden zur Erhöhung der Stabilität entwickelt. Zeolithe mit niedrigem Aluminiumgehalt sind in der Regel thermisch und chemisch stabiler.[33] Da die Synthese von Zeolith Y aber nur in einem bestimmten Bereich bei niedrigem Si/Al-Verhältnis gelingt, wurden Methoden zur nachträglichen Entfernung von Aluminium ausgearbeitet (siehe Dealuminierung).
Barrer und Makki berichteten 1964 erstmals über eine post-synthetische Methode, um Zeolithe (Clinoptilolit) durch Behandlung mit einer Mineralsäure gezielt zu dealuminieren.[36] McDaniel und Maher berichteten 1967 erstmals über die Herstellung eines stabilen Y-Zeolithes, den sie „ultrastabil“ nannten.[30][37] Dieses Material wurde durch Kalzinierung der Ammoniumform (NH4Y) bei über 500 °C dargestellt, wodurch eine Dealuminierung eingeleitet wurde. Kerr zeigte 1967, dass das gebildete Wasser während der Dehydroxylierung von HY eine entscheidende Rolle bei der Ultrastabilisierung spielt.[5] Wenn HY bei 650 °C bis 750 °C unter Spülung mit Inertgas kalziniert wurde, ergab sich ein Zeolith mit niedriger Stabilität. Durch die Spülung mit Inertgas wurde das Wasser ausgetragen, das durch die Abspaltung von Hydroxygruppen gebildet wurde. Wenn dagegen die Kalzinierung bei 700 °C bis 800 °C ohne Inertgasspülung, d. h. in Gegenwart des gebildeten Wassers, durchgeführt wurde, war das Produkt (USY) stabiler. Bei anschließender thermischer Belastung bis 1000 °C blieb es kristallin.
La-ausgetauschte Y-Zeolithe
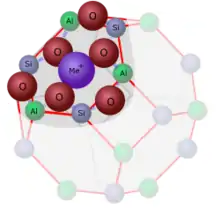
Y-Zeolithe werden zur Erhöhung der Stabilität und Verbesserung der katalytischen Eigenschaften mit Metallen der seltenen Erden, hauptsächlich mit Lanthan und in deutlich geringerem Umfang mit Cer, versetzt (siehe Ionentausch). Oberhalb von 10 Gew.% Gehalt an seltenen Erdmetallen nimmt die Verbesserung der Oktanzahl beim Cracken nicht mehr signifikant zu. Y Zeolithe mit hohem Gehalt an seltenen Erdmetallen werden hauptsächlich zur Maximierung der Benzinausbeute eingesetzt. Y Zeolithe mit niedrigerem Gehalt werden hauptsächlich zur Reduzierung der Verkokung und Bildung von Spaltgasen verwendet. Mit zunehmendem Gehalt an seltenen Erdmetallen nimmt die Oktanzahl des Produktes zu und die Benzinausbeute ab.[15]
La-ausgetauschter USY weist sowohl brönsted- als auch lewissaure Zentren auf.[38] Die Acidität vom Y-Zeolith nimmt durch den Ionentausch mit Lanthan dabei ab, weil ein Teil der Protonen auf der Oberfläche durch Lanthan ersetzt werden. So nimmt die Acidität in der Reihenfolge La,H-Y > USY > La,H-USY > ab.
Die Beständigkeit gegenüber Behandlung mit Wasserdampf bei hohen Temperaturen (540 °C) nimmt durch den Ionentausch mit Lanthan zu. Dieser Test wird zur Simulation der Bedingungen in der FCC-Unit durchgeführt. Durch die Behandlung mit heißem Dampf kann das Gitter vom Zeolith restrukturiert werden, wodurch es zu einer Abspaltung von Wasser und einer Dealuminierung kommen kann. Dies führt zu einem Schrumpfen der Zelleinheit. Die Gegenwart der relativ großen La-Ionen (r = 1,15 A) in den Zeolithkäfigen verhindert das Schrumpfen der Zelleinheit und die Bildung neuer kationischer, aluminiumenthaltender Spezies. Der Zeolith wird dadurch mit zunehmendem La-Gehalt stabiler.
Durch die thermische Behandlung von La-ausgetauschtem Zeolith Y (RE,HY) bilden sich zusätzliche saure Zentren aufgrund der Hydrolyse partiell hydratisierter Lanthanionen.[15] Bei 480 °C kalzinierter RE,HY besitzt nur brönstedsaure Zentren, während die Kalzinierung bei höheren Temperaturen die Brönstedazidität durch Dehydroxylierung abnimmt unter Bildung von lewissauren Zentren. Die hydrothermale Behandlung führt zu Teilhydrolyse der Verbindung zwischen Lanthan und Zeolith und einer partiellen Dealuminierung des Gerüstes mit der zugehörigen Abnahme der Gesamtazidität und Elementarzellengröße.
Xu et al.[39] konnten durch Aufnahmen mit dem Rastertransmissionselektronenmikroskop nachweisen, dass in partiell ausgetauschtem (noch Na-haltigem) LaY die Lanthanionen hauptsächlich als einzelne Kationen vorlagen, dass aber in einem Verhältnis von 1:4 auch Lanthanpaare vorhanden sind, die vermutlich über ein Sauerstoffatom verbrückt sind.
Abriebfestigkeit
Die Abriebfestigkeit des fertigen Katalysators ist aus verschiedenen Gründen wichtig:
- Sie kann die Menge an Katalysator bestimmen, die in der FCC-Unit ständig ersetzt werden muss.
- Sie kann die Fluidisiereigenschaften des Katalysators in der FCC-Unit beeinflussen, da sich die Größenverteilung der Partikel ändert.
- Sie bestimmt die Menge an Feinpartikeln, die aus der FCC-Unit emittiert werden.[15]
Die Abriebfestigkeit wird von der Matrix und vom Zeolithgehalt bestimmt. Allgemein nimmt die Abriebfestigkeit mit zunehmendem Zeolithgehalt ab, was bei Gehalten von über 35 % Zeloth kritisch werden kann.[15]
Porenvolumen und Porenverteilung
Die Porosität des Katalysators wird entscheidend durch die Zusammensetzung, Herstellmethode und hydrothermale Behandlung geprägt. Sie ist eine wichtige Größe, die die katalytischen Eigenschaften des Katalysators beeinflusst. Bei überwiegend kleinen Poren (< 100 Å) besteht die Gefahr einer Verstopfung durch Verkokung.[15] Zu große Makroporen (> 200 Å) werden häufig mit einer zu geringen spezifischen Oberfläche in Verbindung gebracht, was die katalytische Aktivität und die Abriebfestigkeit reduziert. Ein idealer Katalysator enthält eine ausgewogene Verteilung von Mikro- und Makroporen. Während die Mikroporen eine genügend hohe katalytische Aktivität sichern, erlauben die Makroporen eine ausreichend schnelle Diffusion der Reaktionspartner durch den Katalysator. Durch die Verwendung von Tensiden als Template gelingt die Ausbildung mesostrukturierter Poren im Bereich von 2 bis 50 nm.[40] Beispielsweise wird der Zeolith zunächst mit Citronensäure und anschließend mit Cetyltrimethylammoniumbromid (CTAB) und Natronlauge behandelt. Eingeschlossenes CTAB wird durch Kalzinierung bei 550 °C zunächst verkokt und anschließend durch Einleitung von Luft bei dieser Temperatur verbrannt.[41] Die übrig gebliebenen Leerräume stellen Poren mit relativ definierten Abmessungen dar, durch die größere Moleküle leichter in das Zeolithinnere gelangen und die gebildeten Produkte schneller herausdiffundieren können. Dadurch erreicht man eine bessere Selektivität der Spaltreaktion.
Verwendung
Hauptanwendung
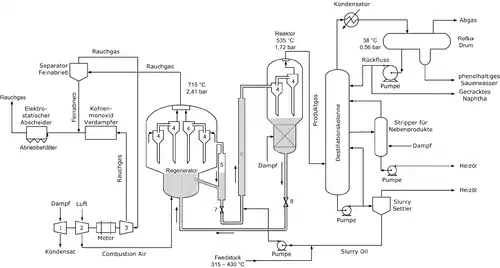
Zeolith Y findet vor allem Anwendung als Crack-Katalysator im FCC-Verfahren zur Umwandlung von Rückständen in Diesel oder Benzin in der Raffinerie. Die zu diesem Zweck hergestellten Mengen liegen im Bereich mehrerer hunderttausend Tonnen im Jahr. Zeolith Y hat Zeolith X als Katalysator verdrängt, da es aufgrund seines höheren Si/Al-Verhältnisses eine höhere Stabilität aufweist und außerdem aktiver ist. Er findet auch im Hydrocracking-Verfahren Verwendung als Träger für Platin/Palladium, um den Gehalt an Aromaten zu erhöhen.
Weitere bedeutende Anwendungen
- Nickel enthaltender Zeolith Y kann als Katalysator in Hydrierreaktionen verwendet werden, z. B. bei der Hydrierung von Kohlenmonoxid in der Fischer-Tropsch-Synthese, bei der Hydrierung von ungesättigten Kohlenwasserstoffen oder bei der Hydrogenolyse von gesättigten Verbindungen.[42]
Weniger bedeutende Anwendungen
- Zeolith Y kann auch als Katalysator für die Herstellung von N-Methylanilin verwendet werden.
- Cobalt-enthaltender Zeolith Y kann ebenfalls für Hydrierreaktionen verwendet werden.[42] Hierbei ist nur Zeolith Y aktiv, welcher mit Co2(CO)8 (siehe Cobaltcarbonylhydrid) imprägniert wird. Ionenausgetauschter Zeolith Y, z. B. aus Cobalt(II)-nitrat, ist nicht hydrieraktiv, da sich das Cobalt in diesem Falle in den Sodalithkäfigen befindet und nicht aus diesen heraus migriert.
Einzelnachweise
- Masters, A.; Maschmeyer, Th.; Zeolites – From curiosity to cornerstone. In: Microporous and Mesoporous Materials 142 (2011) 423-438
- U.S. Patent 3,130,007 Cyrstalline Zeolite Y
- Uytterhoeven, J.B., Christner, L.G., Hall, W.K.; J. Phys. Chem. (1965) 69,2117
- Kerr, G.T., Hydrogen Zeolite Y, Ultrastable Zeolite Y, and Aluminum-Deficient Zeolites, Advances in Chemistry Series, 121/219-229, 1973
- Kerr, G. T.; The Intercrystalline Rearrangement of Constitutive Water in Hydrogen Zeolite Y, J. Phys. Chem. 71 (1967) 4155-4156
- Benesi, H.A., J. Catal. (1967) 8, 368
- Sierka, M., Eichler, U., Datka, J., Sauer, J.; Heterogeinity of Brönsted Acidic Sites in Faujasite Type Zeolites due to Aluminium Content and Framework Structure, J. Phys. Chem. B (1998), 102, 6397-6404
- The Chemical Engineering Zeolite Page
- Kaduk, A.; Crystallopgraphy Reviews, 11 (1) (2005) 1-19
- Breck, D.W., Zeolite Molecular Sieves, 1974, Wiley & Sons
- Marinsky, J.A., Ion Exchange, Volume 2, 169, Dekker Inc.
- „Synthese und katalytische Charakterisierung von Zeolith Y mit unterschiedlicher Kristallgröße“, Dissertation von Christine Berger
- Karami, D.; Rohani, S.; Synthesis of pure zeolite Y using soluble silicate, a two-level factorial experimental design, Chem. Eng. and Proc. 28 (2009) 1288–1292.
- Sohn, J.R., DeCanio, S.J., Lunsford, J.H., O’Donnell, D.J.; Determination of framework aluminium content in dealuminated Y-type zeolites: a comparison based on unit cell size and wavenumbers of IR bands, Zeolites (1986) Vol. 6, 225-227
- Scherzer, J., Octane-Enhancing, Zeolitic FCC-Catalysts: Scientific and Technical Aspects, Catal. Rev.-Sci. Eng. 31 (3) (1989), 215-354
- Kerr, G.T., Cattanach, J., Wu, E.L.; J. Catal. (1969) 13, 114.
- Scherzer, J.: Dealuminated Faujasite-Type Structures with SiO2/Al2O3 Ratios over 100, Journal of Catalysis, 54, (1978), 285-288
- Haden, W.L., et al.; U.S. Patent 3,663,165 (1972)
- Brown, S.M. et al.; U.S. Patent 4,493,902 (1985)
- Stockwell, D. et al.; U.S. Patent 6,656,347 (2003)
- Wang, J.Q. et al.; New hydrothermal route for the synthesis of high purity nanoparticles of zeolite Y from kaolin and quartz, Microporous and Mesoporous Materials 232 (2016) 76-85
- U.S. Patent Re. 28,629 Ion Exchange of Crystalline Zeolites
- Sato, K., et al., Structural changes of Y zeolites during ion exchange treatment: effects of Si/Al ratio of the starting NaY, Microporous and Mesoporous Materials, 59, (2003), pp. 133-146
- Shiralkar, V.P., Kulkarni, S.B., Thermal and structural properties of rare earth exchanged zeolites, Journal of Thermal Analysis, Vol. 25, (1982), pp. 399-407
- Lee, E.F.T., Rees, V.C., Effect of calcination on location and valency of lanthanum ions in zeolite Y, Zeolites, 1987, Vol. 7, pp. 143-147
- Schuessler, F. et al.: Nature and Location of Cationic Lanthanum Species in High Alumina Containing Faujasite Type Zeolites, J. Phys. Chem. C, 2011, 115, 21763 – 21776
- Howard, S., The ion exchange properties of zeolites, III. Rare earth ion exchange of synthetic faujasites, Journal of Colloid and Interface Science, Vol. 28, No. 2, October 1968
- Sulikowski, B. et al.: Solid-state ion exchange in zeolites: Part 8: Interaction of Lanthanum(III) chloride with zeolites under anhydrous conditions, Zeolites (19), 1997, 395-403
- Beyer, H.K.: Dealumination Techniques for Zeolites, Molecular Sieves – Post-Synthesis Modification I, Springer Verlag, 2002, S. 204–255
- van Bekkum, H., et al., Introduction to Zeolite Science and Practice, 2nd Completely Revised and Expanded Edition, Elsevier 2001
- Sievers, C., et al.,Stability of Zeolites in Hot Liquid Water, J. Phys. Chem. C, 2010, 114, 19582-19595
- Kerr, G.T., J. Phys. Chem. 71 (1967), 4155
- Silaghi, M.-C., Chizallet, C., Raybaud, P.; Challenges on molecular aspects of dealumination and desilication of zeolites, Microporous and Mesoporous Materials, 191 (2014) 82-96
- US-Patent 3,506,400 (1970)
- Bremer, H., Morke, W., Schodel, R., Vogt, F.; Adv. Chem. Ser., 121, 249 (1973)
- Barrer, R M., Makki, M.B.: Molecular Sieve Sorbents from Clinoptilolit. In: Canadian Journal of Chemistry. 42 (6), 1964, S. 1481-1487, doi:10.1139/v64-223.
- McDaniel, C.V., Maher, P.K.; Molecular Sieves, in: r.M. Barrer (Ed.), Society of Chemical Industry, London, pp. 186-195
- Scherzer, J., Bass, J.L.: Ion Exchanged Ultrastable Y Zeolites, I. Formation and Structural Characterization of Lanthanum-Hydrogen Exchanged Zeolites, Journal of Catalysis, 46 (1977), 100-108
- Xu, P. et al.: Imaging individual lanthanum atoms in zeolite Y by scanning electron microscopy: Evidence of lanthanum pair sites, Microporous and Mesoporous Materials, 213 (2015) 95-99
- García-Marínez, J., Li, K., Krishnaiah, G.; A mesostructured Y zeolite as a superior FCC catalyst – from lab to refinery, Chem. Commun. 2012, 48, 11841-11843
- U.S: Patent 20110118107
- Coughlan, C., Ó Domhnaill, C.; Catalysis on cobalt-containing sodium zeolite Y. In: Topics in Catalysis, 1 (1994), 163-167
Literatur
- Subhash Bhatia: Zeolite Catalysis. Principles and Applications. CRC Press Inc., Boca Raton FL 1990, ISBN 0-8493-5628-8.
- F. Ramoa Ribeiro u. a. (Hrsg.): Zeolites. Science and Technology. Proceedings of the NATO Advanced Study Institute on Zeolites, Science and Technology, Alcabideche, Portugal, May 1–12, 1983. Martinus Nijhoff Publishers u. a., The Hague u. a. 1984, ISBN 90-247-2935-1 (NATO ASI series: E: Applied sciences 80).
Weblinks
- „Synthese und katalytische Charakterisierung von Zeolith Y mit unterschiedlicher Kristallgröße“, Dissertation von Christine Berger
- The Chemical Engineering Zeolite Page
- James A. Kaduk, Johan Faber: Crystal Structure of Zeolite Y as Function of Ion Exchange (PDF; 3,1 MB). The Rigaku Journal, Vol. 12, No. 2, 1995