Michel Eichelbaum
Michel Eichelbaum (* 19. Mai 1941 in Leipzig) ist ein deutscher Internist und Klinischer Pharmakologe. Er ist besonders bekannt durch seine Arbeiten über den Einfluss von Genmutationen auf die Pharmakokinetik von Arzneistoffen.

Leben
Seine Eltern waren der Kaufmann Gustav Eichelbaum und seine Frau Emma, geb. Michel. Der Sohn Michel bestand 1959 an der Leibniz-Oberschule in Leipzig die Abiturprüfung. Im selben Jahr floh er mit der Mutter und zwei jüngeren Brüdern über Berlin (West) nach Westdeutschland. 1960 legte er in Stuttgart die westdeutsche Ergänzungsprüfung zum DDR-Abitur ab. Anschließend studierte er bis zum Staatsexamen 1966 in Heidelberg Medizin. 1968 wurde er mit einer bei Hans J. Dengler (1925–1997)[1] in Heidelberg angefertigten Dissertation „Aufnahme, Verteilung, Ausscheidung und Metabolismus von 14C-Prenylamin“ zum Dr. med. promoviert. Als Dengler 1968 Direktor der Medizinischen Poliklinik der Universität Gießen wurde, folgte Eichelbaum ihm, ebenso, als Dengler 1973 den Lehrstuhl für Innere Medizin der Universität Bonn übernahm. Dort habilitierte er sich 1976 mit einer Arbeit „Ein neuentdeckter Defekt im Arzneimittelmetabolismus des Menschen: Die fehlende N-Oxidation des Sparteins“
1970 und 1971 arbeitete er bei Bernard Beryl Brodie (1907–1989) und James R. Gillette (1928–2001) im Laboratory of Chemical Pharmacology des National Heart, Lung and Blood Institute der National Institutes of Health in Bethesda, 1973 und 1974 bei Folke Fritz Gustaf Sjöqvist (1933–2020) am Department of Clinical Pharmacology des Karolinska-Instituts in Solna bei Stockholm.
1985 wurde er als Nachfolger von Jürgen C. Fröhlich (* 1939) Direktor des Dr. Margarete Fischer-Bosch-Instituts für Klinische Pharmakologie am Robert-Bosch-Krankenhaus Stuttgart. 1996 übernahm er zusätzlich den damals neu geschaffenen Lehrstuhl für Klinische Pharmakologie der Universität Tübingen. Das Wintersemester 1995–1996 verbrachte er als Visiting Professor am Department of Clinical and Experimental Pharmacology der University of Adelaide bei Felix Bochner (* 1939) und Andrew Somogyi.[2][3]
Seit 2007 emeritiert, arbeitet Eichelbaum weiter an seinem ehemaligen, seit 2007 von seinem Schüler Matthias Schwab (* 1963) geleiteten Institut am Robert-Bosch-Krankenhaus. Er hat einen Sohn und eine Tochter.
Forschung
Klinische Pharmakologie
Schon in Eichelbaums Heidelberger Dissertation deutete sich seine weitere Forschung an. Es ging um das Schicksal des damals bei Koronarer Herzkrankheit verwendeten Prenylamins (Segontins®) im Körper, das heißt um die Pharmakokinetik dieses Stoffes. Er untersuchte sie für die Dissertation im Tierversuch,[4] später, in Gießen, beim Menschen. Die Pharmakokinetik, vor allem beim Menschen, wurde sein Hauptthema. Dabei behielt er die Pharmakodynamik, also die Wirkung der Arzneistoffe, stets im Blick; sie wird ja durch die Pharmakokinetik, also zum Beispiel die Elimination, mitbestimmt. So wurde Eichelbaum – wie sein erster und wichtigster akademischer Lehrer Dengler – Internist und Klinischer Pharmakologe. Als solcher untersuchte er ein breites Spektrum von Themen, darunter das Antiepileptikum Carbamazepin und das Antiarrhythmikum Propafenon. Hervorzuheben sind Ergebnisse zur Genetik der Cytochrom P450-Enzyme, zur Rolle der Chiralität in der Pharmakologie und zu Fremdstoff-Transportern.
Spartein-Debrisoquin-Polymorphismus
Noch in Gießen begannen Arbeiten über das Besenginster-Alkaloid Spartein, das damals als Antiarrhythmikum und Mittel zur Geburtseinleitung erprobt wurde. Sie führten zu einer folgenreichen Entdeckung, über die Eichelbaum und seine Koautoren erstmals bei einer Tagung der Deutschen Pharmakologischen Gesellschaft 1975 berichteten (aus dem Englischen): „Bei der Analyse der Pharmakokinetik von Spartein fanden wir eine kleine Zahl anscheinend gesunder Personen, die praktisch unfähig waren, Spartein zu metabolisieren. ... Diese ‚Nicht-Metabolisierer‘ wandelten Spartein nicht in die entsprechenden ... Oxidationsprodukte um. Von mehr als 100 Probanden zeigten 4 die Abweichung. Bei diesen 4 wurden mehr als 90 % einer Spartein-Dosis unverändert als Spartein ausgeschieden, bei allen anderen nur 15–30 % der Dosis. Die Spartein-Plasmaspiegel waren bei den Nicht-Metabolisierern zwei- bis viermal höher als bei den ‚Metabolisierern‘. ... Nur die Nicht-Metabolisierer, nicht die Metabolisierer erfuhren unerwünschte Wirkungen wie Sehstörungen, Kopfschmerz und Schwindel.“[5]
Zwei Jahre später berichtete eine Londoner Arbeitsgruppe über einen anderen interindividuellen Unterschied bei einer Arzneistoffoxidation: Das Antihypertensivum Debrisoquin wurde bei 91 von 94 Personen ausgiebig oxidiert, bei den 3 anderen kaum. Der Unterschied war genetisch bedingt.[6] Weitere drei Jahre später, 1980, brachten Eichelbaum und seine Mitarbeiter die beiden Anomalien zusammen: Es handelte sich bei dem Defekt im Spartein-Metabolismus und dem Defekt im Debrisoquin-Metabolismus um genetisch bedingte Defekte in demselben Enzymsystem, einen Spartein-Debrisoquin-Polymorphismus.[7][8]
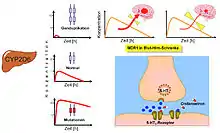
Schon in den 1950er Jahren waren genetische Variationen von arzneistoffmetabolisierenden Enzymen gefunden worden. Dabei hatte es sich aber um relativ spezielle Enzyme gehandelt, zum Beispiel die Pseudocholinesterase, die zwei Muskelrelaxantien spaltet. Das für die Spartein- und Debrisoquinoxidation verantwortliche Enzym aber gehörte, wie Eichelbaum schon in seiner Habilitationsschrift gezeigt hatte, zum Cytochrom P450-System, dem wichtigsten, die allermeisten Fremdstoffe (durch Oxidation) abbauenden Enzymsystem.
In der Folge wurden die Auswirkung des Defekts auf die Metabolisierung weiterer Arzneistoffe und seine Häufigkeit bei verschiedenen ethnischen Gruppen erforscht. Das Gen für das verantwortliche P450-Enzym wurde auf den langen Arm des Chromosoms 22 lokalisiert[9] und von einer US-amerikanisch-schweizerischen Forschergruppe kloniert.[10] Das Enzym erhielt den Namen Cytochrom P450 2D6 (CYP 2D6). Auch zahlreiche Mutationen, die zu Verminderung (oder auch Steigerung) der Enzymaktivität führten, wurden identifiziert,[11] bis heute (2011) mehr als 100. Es gibt danach normale Metabolisierer (extensive metabolizers), defiziente Metabolisierer (poor metabolizers) oder auch extrem schnelle Metabolisierer (ultrarapid metabolizers). Die Bedeutung rührt einerseits von der großen Zahl der betroffenen Menschen her, bei Europäern etwa 15 % defiziente Metabolisierer, andererseits von der großen Zahl der Arzneistoffe, die ausschließlich oder teilweise durch CYP 2D6 metabolisiert werden, mehr als 50 – darunter das Propafenon.[12][13] So trägt, ein Beispiel von vielen, CYP 2D6 – und damit sein Polymorphismus – zur Metabolisierung von Tamoxifen bei und beeinflusst dadurch dessen Wirksamkeit bei Brustkrebs.[14][15]
Verapamil: stereoselektive Pharmakokinetik
Verapamil war Ende der 1960er Jahre von Albrecht Fleckenstein als ein Calciumantagonist erkannt worden und wird bis heute bei Herz-Kreislauferkrankungen häufig angewandt. Bald fiel auf, dass die übliche Dosis für intravenöse Gabe, 5–10 mg, bei oraler Gabe wirkungslos war und sogar eine fünf- bis zehnfach höhere Dosis oral nur unzuverlässig wirkte. Um den Grund zu finden, entwickelte Eichelbaums Gruppe ein gaschromatographisch-massenspektrometrisches Verfahren zur Messung der Substanz und ihrer Metaboliten. Es stellte sich heraus, dass Verapamil zwar vollständig aus dem Darm resorbiert, anschließend aber in der Darmwand und Leber großenteils abgebaut wurde, das Herz also durch diesen First-Pass-Effekt, den Abbau bei der ersten Passage durch Darmwand und Leber, nicht erreichte.[16] Damit war eine Frage beantwortet.
Die nächste stellte sich mit dem Paradoxon, dass eine bestimmte Konzentration von Verapamil im Blut, erreicht durch orale Gabe, das Herz weniger beeinflusste als dieselbe Konzentration, erreicht durch intravenöse Gabe.[17] „We then realised that verapamil had a chiral centre and was administered as a racemate – Da erinnerten wir uns, dass Verapamil ein chirales Molekül war und als Racemat verwendet wurde.“[18] Die Bonner Forscher hatten, wie damals nicht anders möglich, nur die Gesamtkonzentration von „(R)- + (S)-Verapamil“ gemessen. Weil zudem bekannt war, dass das (S)-Enantiomer der eigentliche Wirkstoff war, jedenfalls am Herzen viel stärker wirkte als das (R)-Enantiomer, stellten sie die Hypothese auf, die Leber baue beim First-Pass-Effekt vorwiegend das (S)-Enantiomer ab. Dann sollte das Blut bei einer bestimmten Gesamtkonzentration von „(R)- + (S)-Verapamil“, erreicht durch orale Gabe, weniger des wirksamen (S)-Verapamils enthalten als bei derselben Gesamtkonzentration, erreicht durch intravenöse Gabe.
Man konnte damals die Enantiomere nicht trennen. Der Weg zur Testung der Hypothese musste neu erdacht werden. Er bestand in der Markierung eines der beiden Isomere mit dem stabilen Wasserstoff-Isotop Deuterium. Das deuterierte Enantiomer konnte dann dank seiner größeren Masse im Massenspektrometer von dem nicht deuterierten getrennt werden. 1984 lagen die Resultate vor. Wirklich unterlag (S)-Verapamil einem stärkeren First-Pass-Effekt als (R)-Verapamil, und nach gleicher oraler Dosis war die Konzentration von (S)-Verapamil im Blut viel geringer als die Konzentration von (R)-Verapamil, eine stereoselektive Metabolisierung mit erheblichen Folgen für die Wirkung des Stoffes.[19] (R)- und (S)-Verapamil unterscheiden sich auch in ihrer Bindung an Plasmaproteine.[20] Die stereoselektive Pharmakokinetik wurde ein eigenes Forschungsgebiet.[21] Auch Spartein wird stereoselektiv metabolisiert,[22] und die Enantiomere des Propafenons unterscheiden sich in Metabolisierung wie Wirkung.[23] Die Publikation von 1984 wurde 2004 in einem Sonderheft Citation classics in the British Journal of Clinical Pharmacology 1974–2003 nachgedruckt, mit dem Kommentar Eichelbaums, er halte sie für eine seiner besten.[18]
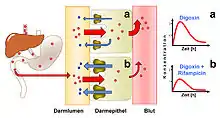
Digoxin und Rifampicin: Transporter
Die Pharmakokinetik wird durch Transporter mitbestimmt, die Fremdstoffe durch Zellmembranen befördern. Ein wichtiger Transporter ist das P-Glykoprotein. Es schleust unter Verbrauch von Adenosintriphosphat Fremdstoffe aus Zellen heraus. So werden zum Beispiel Zytostatika aus Krebszellen herausgeschleust und die Zellen dadurch Zytostatika-resistent – diese Wirkung führte 1976 zur Entdeckung und führte auch zu dem Alternativnamen Multidrug-Resistance-Protein 1 (MDR1; auch ABCB1). Verapamil-Racemat, so wusste man seit 1981, hemmte den Auswärtstransport. Seiner Anwendung bei Tumoren standen aber die (in diesem Fall unerwünschten) Herz-Kreislauf-Wirkungen entgegen. Eichelbaum mit seiner Erfahrung in stereoselektiver Pharmakologie veranlasste deshalb einen Vergleich der Wirkung von (R)- und (S-)-Verapamil auf Krebszellen: Beide steigerten die Wirkung von Zytostatika, und zwar gleich stark. Anders als das Racemat, so die Folgerung, könnte das an Herz und Kreislauf wenig wirksame (R)-Enantiomer ein brauchbares Hilfsmittel in der Krebs-Chemotherapie werden[24] – eine Hoffnung, die sich letztlich nicht erfüllte.
P-Glykoprotein kommt auch in gesunden Zellen vor und hat mehrere pharmakokinetische Funktionen. Zum Beispiel schränkt es die Resorption mancher Arzneistoffe wie des Herzglykosids Digoxin aus dem Darm ein, indem es sie ins Darmlumen zurück pumpt. Daraus resultierte, so entdeckten die Stuttgarter Forscher 1999, eine neuartige Arzneistoff-Wechselwirkung: Einnahme des Antibiotikums Rifampicin zusätzlich zu Digoxin verminderte dessen Konzentration im Blut, indem es den Gehalt der Darmschleimhaut an P-Glykoprotein erhöhte und so den Rücktransport des Digoxins weiter verstärkte.[25] Der Mechanismus der P-Glykoprotein-Vermehrung – Erhöhung seines Transkription des Gens – wurde geklärt,[26] andere Stoffe, zum Beispiel aus dem Johanniskraut, wirkten wie Rifampicin, und Analoges wurde an anderen Transportern beobachtet. Die Induktion von Arzneistoff-metabolisierenden Enzymen hatte ein Gegenstück in der Induktion von Transportern gefunden.[27]
In Zusammenarbeit mit einer Gruppe an der Charité in Berlin und einem biotechnologischen Unternehmen suchte Eichelbaum Gruppe Ende der 1990er Jahre systematisch nach Mutationen im Gen des P-Glykoproteins. Man fand fünfzehn, und eine davon (C3435T) hatte pharmakokinetische Folgen: Bei homozygoten Trägern der Mutation war die Menge an P-Glykoprotein im Darm vermindert und nach Digoxin-Einnahme die Konzentration des Glykosids im Blut erhöht.[28] 25 Jahre nach der Entdeckung des CYP 2D6-Polymorphismus war dies der erste klinisch relevante Polymorphismus eines Transporters. Die Publikation hat viel weitere Forschung angeregt, und die allgemeine medizinische Bedeutung der Mutation ist nicht klar.[29] Sie scheint das Risiko einer Erkrankung an Nierentumoren, Colitis ulcerosa und der Parkinson-Krankheit zu erhöhen[30] und beeinflusst den Behandlungserfolg bei Colitis ulcerosa.[31]
Wissensvermittlung
Eichelbaum hat seine Ergebnisse und Kenntnisse den Gesundheitsberufen und der allgemeinen Öffentlichkeit schreibend und organisierend zu vermitteln versucht. Von 1987 bis 1990 war er Präsident der Deutschen Gesellschaft für Klinische Pharmakologie und von 2000 bis 2003 Vizepräsident der Deutschen Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie. Er ist Mitglied der Arzneimittelkommission der deutschen Ärzteschaft und arbeitet an deren jetzt in 22. Auflage vorliegenden Arzneiverordnungen – Empfehlungen zur rationalen Pharmakotherapie mit.
Er gibt klinisch-pharmakologische Fachzeitschriften (mit) heraus wie das European Journal of Clinical Pharmacology und Pharmacogenetics and Genomics. In Übersichtsartikeln für Ärzte hat er die Bedeutung der Pharmakogenetik für eine individualisierte Medizin betont.[32][33] Mit Matthias Schwab hat er sein Fach in Lehrbuchartikeln dargestellt.[34]
Im Jahr 2007 setzte er sich in einer internationalen Gruppe von Wissenschaftlern für ein Netzwerk zur Vermeidung schwerer Arzneimittel-Nebenwirkungen (SADR) ein: „When good drugs go bad. … It is unthinkable that selecting drugs for individual patients remains an empirical exercise. … A global SADR network will not be cheap, but it will be a fraction of the current cost of SADRs to the healthcare system. – Wenn gute Arzneimittel zu schlechten werden. … Es wäre unverantwortlich, die Auswahl von Arzneimitteln für individuelle Patienten ungeprüfter Meinung zu überlassen. … Ein globales SADR-Netzwerk wäre nicht billig, würde aber nur einen Bruchteil der gegenwärtigen Kosten für SADR verursachen.“[35]
Anerkennung
Im Jahre 1976 erhielt Eichelbaum den Paul-Martini-Preis der gleichnamigen Stiftung und 1983 die gleichnamige Medaille. 1988 wurde er gemeinsam mit Konrad Beyreuther mit dem Robert Pfleger-Forschungspreis ausgezeichnet. 1991 ehrte ihn die Deutsche Gesellschaft für Pharmakologie und Toxikologie mit der Rudolf-Buchheim-Vorlesung. 2001 wurde er Mitglied der Deutschen Akademie der Naturforscher Leopoldina,[36] 2003 der Akademie der Wissenschaften und der Literatur in Mainz[37]. 2008 verlieh ihm die Deutsche Gesellschaft für Experimentelle und Klinische Pharmakologie und Toxikologie ihre höchste wissenschaftliche Auszeichnung, die Schmiedeberg-Plakette.
Einzelnachweise
- M. Eichelbaum und K. von Bergmann: Prof. Dr.med. Hans J. Dengler. In: DGPT-Forum Heft 23, 1998, S. 9–10.
- Wilhelm Kirch: Oswald-Schmiedeberg-Medaille der DGPT für Prof. Eichelbaum. In: Biospektrum 14, 2008, S. 309. Abgerufen am 17. Januar 2013.
- Eichelbaum auf der Internetseite der Arzneimittelkommission der deutschen Ärzteschaft. (Memento des Originals vom 25. Juli 2014 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. Abgerufen am 17. Januar 2013.
- H. J. Dengler und M. Eichelbaum: Untersuchungen zum Verhalten von C14-D,L-Prenylamin im Organismus. In: Naunyn-Schmiedebergs Archiv für Pharmakologie und Experimentelle Pathologie. 260, 1968, S. 105–106. doi:10.1007/BF00537915.
- M. Eichelbaum, N. Spannbrucker, H. J. Dengler: N-oxidation of sparteine in man and its interindividual differences. In: Naunyn-Schmiedebergs Archives of Pharmacology. 287, 1975, S. R 94.
- A. Mahgoub, J. R. Idle, L. G. Dring, R.Lancaster, R. L. Smith: Polymorphic hydroxylation of debrisoquine in man. In: The Lancet. 310, 1977, S. 584–586. doi:10.1016/S0140-6736(77)91430-1.
- L. Bertilsson, H. J. Dengler, M. Eichelbaum, H. U. Schulz: Pharmacogenetic covariation of defective N-Oxidation of sparteine and 4-hydroxylation of debrisoquine. In: European Journal of Clinical Pharmacology. 17, 1980, S. 153–155. doi:10.1007/BF00562624.
- M. Eichelbaum, L. Bertilsson, J. Säwe, C. Zekorn: Polymorphic oxidation of sparteine and debrisoquine: related pharmacogenetic entities. In: Clinical Pharmacology and Therapeutics. 31, 1982, S. 184–186. doi:10.1038/clpt.1982.29.
- M. Eichelbaum, M. P. Baur, H. J. Dengler, B. O. Osikowska-Evers, G. Tieves, C. Zekorn und C. Rittner: Chromosomal assignment of human cytochrom P-450 (debrisoquin/sparteine type) to chromosome 22. In: British Journal of Clinical Pharmacology. 23, 1987, S. 455–458. PMC 1386095 (freier Volltext).
- Frank J. Gonzalez, Radek C. Skodat, Shioko Kimura, Morio Umeno, Ulrich M. Zanger, Daniel W. Nebert, Harry V. Gelboin, James P. Hardwick und Urs A. Meyer: Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. In: Nature. 331, 1988, S. 442–446. doi:10.1038/331442a0.
- Alan C Gough, John S. Miles, Nigel K. Spurr, Julie E. Moss, Andrea Gaedigk, Michel Eichelbaum und C. Roland Wolf: Identification of the primary gene defect at the cytochrome P450 CYP2D locus. In: Nature. 347, 1990, S. 773–776. doi:10.1038/347773a0.
- Ulrich M. Zanger, Sebastian Raimundo, Michel Eichelbaum: Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. In: Naunyn-Schmiedeberg’s Archives of Pharmacology. 369, 2004, S. 23–37. doi:10.1007/s00210-003-0832-2.
- M. Eichelbaum, M. Schwab: Wirkungen des Organismus auf Pharmaka: allgemeine Pharmakokinetik. In: K. Aktories, U. Förstermann, F. Hofmann, K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009, Seite 36–64, hier S. 63. ISBN 978-3-437-42522-6
- Werner Schroth, Matthew P. Goetz, Ute Hamann, Peter A. Fasching, Marcus Schmidt, Stefan Winter, Peter Fritz, Wolfgang Simon, Vera J. Suman, Matthew M. Ames, Stephanie L. Safgren, Mary J. Kuffel, Hans Ulrich Ulmer, Julia Boländer, Reiner Strick, Matthias W. Beckmann, Heinz Koelbl, Richard M. Weinshilboum, James N. Ingle, Michel Eichelbaum, Matthias Schwab, Hiltrud Brauch: Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. In: Journal of the American Medical Association. 302, 2009, S. 1429–1436. doi:10.1001/jama.2009.1420.
- T. E. Mürdter, W. Schroth, L. Bacchus-Gerybadze, S. Winter, G. Heinkele, W. Simon, P. A. Fasching, T. Fehm, M. Eichelbaum, M. Schwab, H. Brauch: Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and phase II enzymes on their concentration levels in plasma. In: Clinical Pharmacology and Therapeutics. 89, 2011, S. 708–717. doi:10.1038/clpt.2011.27.
- Michael Schomerus, Berthold Spiegelhalder, Barbara Stieren, Michel Eichelbaum: Physiological disposition of verapamil in man. In: Cardiovascular Research. 10, 1976, S. 605–612. doi:10.1093/cvr/10.5.605.
- M. Eichelbaum, P. Birkel, E. Grube, U. Gütgemann, A. Somogyi: Effects of verapamil on P-R-intervals in relation to verapamil plasma levels following single i. v. and oral administration and during chronic treatment. In: Klinische Wochenschrift. 58, 1980, S. 919–925. doi:10.1007/BF01477049.
- Michel Eichelbaum: Author’s commentary. In: British Journal of Clinical Pharmacology. 58, 2004, S. S804–S805. doi:10.1111/j.1365-2125.2004.02300.x.
- B. Vogelgesang, H. Echizen, S. Schmidt, M. Eichelbaum: Stereoselective first-pass metabolism of highly cleared drugs: studies of the bioavailability of L- and D-verapamil examined with a stable isotope technique. In: British Journal of Clinical Pharmacology. 18, 1984, S. 733–740. PMC 1463564 (freier Volltext)., abgerufen am 18. Oktober 2011.
- Annette S. Gross, Barbara Heuer, Michel Eichelbaum: Stereoselective protein binding of verapamil enantiomers. In: Biochemical Pharmacology. 24, 1988, S. 4623–4627. doi:10.1016/0006-2952(88)90330-9.
- A.S. Gross, A. Somogyi, M. Eichelbaum: Stereoselective drug metabolism and drug interactions. In: Michel Eichelbaum, Bernard Testa, Andrew Somogyi: Stereochemical Aspects of Drug Action and Disposition. Handbook of Experimental Pharmacology 153, S. 313–339. Berlin, Heidelberg, Springer-Verlag 2003. ISBN 3-540-41593-9
- T. Ebner, C. O. Meese, M. Eichelbaum: Regioselectivity and stereoselectivity of the metabolism of the chiral quinolizidine alkaloids sparteine and pachycarpine in the rat. In: Xenobiotica. 21, 1991, S. 847–857. doi:10.3109/00498259109039524.
- Heyo K. Kroemer, Christian Funck-Brentano, David J. Silberstein, Alastair J. J. Wood, Michel Eichelbaum, Raymond L. Woosley, Dan M. Roden: Stereoselective disposition and pharmacologic activity of propafenone enantiomers. In: Circulation. 79, 1989, S. 1068–1076. doi:10.1161/01.CIR.79.5.1068.
- K. Häußermann, B. Benz, V. Gekeler, K. Schumacher, M. Eichelbaum: Effects of verapamil enantiomers and major metabolites on the cytotoxicity of vincristine and daunomycin in human lymphoma cell lines. In: European Journal of Clinical Pharmacology. 40, 1991, S. 53–59. doi:10.1007/BF00315139.
- Bernd Greiner, Michel Eichelbaum, Peter Fritz, Hans-Peter Kreichgauer, Oliver von Richter, Johannes Zundler, Heyo K. Kroemer: The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. In: The Journal of Clinical Investigation. 104, 1999, S. 147–153. doi:10.1172/JCI6663.
- Anke Geick, Michel Eichelbaum, Oliver Burk: Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. In: The Journal of Biological Chemistry. 276, 2001, S. 14581–14587. doi:10.1074/jbc.M010173200.
- D. Rosskopf, H. K. Kroemer, W. Siegmund: Pharmakokinetische Probleme in der Praxis – Rolle von Arzneimitteltransportern. In: Deutsche Medizinische Wochenschrift. 134, 2009, S. 345–356. doi:10.1055/s-0028-1124005.
- S. Hoffmeyer, O. Burk, O. von Richter, H.P. Arnold, J. Brockmöller, A. Johne, I. Cascorbi, T. Gerloff, I. Roots, M. Eichelbaum, U. Brinkmann: Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glykoprotein expression and activity in vivo. In: Proceedings of the National Academy of Sciences. 97, 2000, S. 3473–3478. doi:10.1055/s-0028-1124005.
- Ann K. Daly: Pharmacogenetics and human genetic polymorphisms. In: Biochemical Journal. 429, 2010, S. 435–449. doi:10.1042/BJ20100522.
- Matthias Schwab, Michel Eichelbaum und Martin F. Fromm: Genetic polymorphisms of the human MDR1 drug transporter. In: Annual Review of Pharmacology and Toxicology. 43, 2003, S. 285–307. doi:10.1146/annurev.pharmtox.43.100901.140233.
- K. R. Herrlinger, H. Koc, S. Winter, A. Teml, E. F. Stange, K. Fellermann, P. Fritz, M. Schwab, E. Schaeffeler: ABCB1 single-nucleotide polymorphisms determine tacrolimus response in patients with ulcerative colitis. In: Clinical Pharmacology and Therapeutics. 89, 2011, S. 422–428. doi:10.1038/clpt.2010.348.
- M. Eichelbaum, M. Schwab: Pharmakogenetik. In: Monatsschrift Kinderheilkunde. 153, 2005, S. 741–750. doi:10.1007/s00112-005-1199-x.
- Michel Eichelbaum, Magnus Ingelman-Sundberg und William E. Evans: Pharmacogenomics and individualized drug therapy. In: Annual Review of Medicine. 57, 2011, S. 119–137. doi:10.1146/annurev.med.56.082103.104724.
- M. Eichelbaum, M. Schwab: Wirkungen des Organismus auf Pharmaka: allgemeine Pharmakokiketik und Arzneistoffkonzentration im Organismus in Abhängigkeit von der Zeit: Pharmakokinetik im engeren Sinn. In: K. Aktories, U. Förstermann, F. Hofmann, K. Starke: Allgemeine und spezielle Pharmakologie und Toxikologie. 10. Auflage, München, Elsevier GmbH 2009, Seite 36–79. ISBN 978-3-437-42522-6
- Kathleen M. Giacomini, Ronald M. Krauss, Dan M. Roden, Michel Eichelbaum, Michael R. Hayden, Yusuke Nakamura: When good drugs go bad. In: Nature. 446, 2007, S. 975–977. doi:10.1038/446975a.
- Mitgliedseintrag von Michel Eichelbaum bei der Deutschen Akademie der Naturforscher Leopoldina, abgerufen am 5. Juli 2016.
- Mitgliedseintrag von Michel Eichelbaum bei der Akademie der Wissenschaften und der Literatur Mainz, abgerufen am 11.10.17