Kristallisation (Polymer)
Die Kristallisation von Polymeren ist bei einigen thermoplastischen Kunststoffen zu beobachten. Hier kommt es beim Erstarren der Schmelze zu einer partiellen Ordnung der Molekülketten im Polymer. Ausgehend von Kristallisationskeimen lagern sich die Molekülketten faltenförmig aneinander und bilden sogenannte Lamellen, welche durch amorphe Bereiche getrennt sind. Die Lamellen bilden Überstrukturen wie z. B. Sphärolithe. Neben dem Erstarren kann eine Kristallisation auch aus einer Lösung erfolgen.
Die Kristallitbildung ist abhängig von den Abkühlbedingungen, den Additiven und Füllstoffen im Polymer sowie den Strömungsbedingungen während des Erstarrens. Auch eine nachträgliche Verstreckung verändert die Anordnung der Moleküle und damit die Eigenschaften des Materials.
Die Kristallisation hat Einfluss auf die optischen, mechanischen, thermischen und chemischen Eigenschaften des Polymers und seine Verarbeitung. Der Kristallisationsgrad ist durch verschiedene analytische Methoden messbar. Die Eigenschaften werden jedoch nicht nur vom Kristallisationsgrad, sondern auch von der Größe der Struktureinheiten oder der Molekülorientierung bestimmt.
Kristallisationsmechanismen
Nach wie vor sind viele Phänomene rund um die Kristallisation von polymeren Werkstoffen nicht endgültig verstanden oder gar nachgewiesen. Verschiedene Modelle wurden durch experimentelle Befunde gestützt und haben sich durchgesetzt:[1]
Kristallitbildung beim Erstarren aus der Schmelze

Polymere sind aus sehr langen Molekülketten aufgebaut. Thermoplastische Polymere zeichnen sich dadurch aus, dass sie sich bei Temperaturerhöhung stark erweichen und schließlich fließfähig werden. Falls kristalline Bereiche vorliegen, schmelzen diese. In der Schmelze sind die Molekülketten dann unregelmäßig in Form von Knäueln angeordnet (Abb. 1), die einander vielfältig durchdringen (Verschlaufung). Bei vielen thermoplastischen Polymeren bleibt diese Unordnung bei der Abkühlung als amorphe Struktur im erstarrten Festkörper erhalten.[2]
Kühlt man hingegen die Schmelze eines teilkristallinen Polymers (eine Untergruppe der Thermoplaste) ab, so bewegen sich die Ketten immer weniger und beginnen sich regelmäßig anzuordnen (Kristallisation). Es kommt zu einer Ausbildung von Ordnungszuständen („Kristalliten“) mit einer typischen Größe von 15–100 nm.[2]
Bei der Kristallisation von Polymeren lagern sich Abschnitte der Molekülketten parallel aneinander. Energetisch am günstigsten wäre, wenn die Moleküle über die gesamte Länge der Molekülkette parallel angeordnet wären. Da die Molekülketten in der Schmelze jedoch als wirre, miteinander verschlungene Knäuel vorliegen, ist diese Ordnung in der Realität nicht oder nur unter sehr hohem Druck erreichbar. Es bilden sich daher Kristallite aus gefalteten Molekülketten (Abb. 1), welche die Grundstrukturen größerer Struktureinheiten wie z. B. Lamellenstrukturen bilden.[2] Die Ordnung ist dabei nicht als vollständig anzusehen. Es können sich an den Faltungsbögen z. B. kleinere oder größere Schlaufen bilden. Auch die Kettenenden können ungeordnet vorliegen. Daneben ist es üblich, dass eine Molekülkette einen Kristallit verlässt und in einen anderen wieder einmündet. Jeder Kristallit besteht daher aus geordneten (kristallinen) und ungeordneten (amorphen) Teilbereichen.[2] Dieses ist auch der Grund, dass selbst in dem Fall, dass das Polymer makroskopisch keine amorphen Bereiche aufweist, ein Polymer-Werkstoff nur als teilkristallin bezeichnet werden kann.
Bislang konnten weder verknäulte Moleküle in der Lösung oder Schmelze, noch gefaltete Molekülketten im festen Polyethylen direkt sichtbar gemacht und fotografisch dokumentiert werden. Für die Richtigkeit des Faltenmodells bei aus der Schmelze erstarrtem Polyethylen gibt es jedoch einen zwingenden Nachweis, indem es gelang, die Lamellen mechanisch durch einen Oberflächenabriss von einer durch langsame Abkühlung aus der Schmelze erstarrten massiven Probe von Niederdruck-Polyethylen (PEHD) mit einer mittleren Molmasse von M = 100 kg/mol zu trennen, im Transmissionselektronenmikroskop sichtbar zu machen und fotografisch zu dokumentieren. Damit ist bewiesen, dass die Bindung zwischen den Lamellen kleiner ist als die Bindung zwischen den Kohlenstoffatomen der Molekülkette im Innern der Lamellen. Die Länge der Moleküle ist dabei um ein Vielfaches größer als die Dicke der Lamellen. Dies entspricht völlig der Erklärung des Begründers des Faltungsmodells, A. Keller: "Wenn die Lamellen isolierte Einzelobjekte sind, wie nach der Kristallisation aus der Lösung, und die Ketten senkrecht oder unter einem großen Winkel zur Grundfläche angeordnet sind, dann ist die Faltung eine direkte Notwendigkeit, da die Ketten nicht anderswo hin gehen können. Dies war die ursprüngliche Basis des Kettenfaltungspostulats von 1957 und diese bleibt jetzt genau so wahr wie sie damals war."[3]
 Kohlenstoffabdruck einer Bruchfläche von PEHD mit anhaftenden Lamellen
Kohlenstoffabdruck einer Bruchfläche von PEHD mit anhaftenden Lamellen Kohlenstoffabdruck einer Bruchfläche von PEHD mit anhaftenden Lamellen
Kohlenstoffabdruck einer Bruchfläche von PEHD mit anhaftenden Lamellen Präparationsschritte für den Nachweis der Lamellentrennung mit dem Elektronenmikroskop
Präparationsschritte für den Nachweis der Lamellentrennung mit dem Elektronenmikroskop
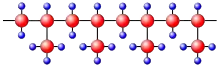
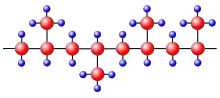
Ob Kunststoffe kristallisieren können, hängt von ihrem molekularen Aufbau ab. Am besten kristallisieren unverzweigte Molekülketten mit keinen oder möglichst wenigen, dafür aber regelmäßig angeordneten Seitengruppen. Beispiele für teilkristalline Polymere sind lineares Polyethylen (PE), Polytetrafluorethylen (PTFE) oder isotaktisches Polypropylen (PP).[2]
Beispiel: Beim isotaktischen Polypropylen sind die CH3-Seitengruppen regelmäßig alle auf einer Seite der Molekülkette angeordnet (Abb. 2a). Damit ist es möglich, dass sich zwei derartige Kettenteile nahezu an allen Positionen aneinanderlagern können. Es gibt jedoch auch Polymere, bei denen die Seitengruppen an verschiedenen Seiten der Kette angebracht sind. Kommt noch zusätzlich eine unregelmäßige Abfolge der Seitenketten hinzu (wie z. B. beim ataktischen Polypropylen in Abb. 2b), so kommt es nur dann zu einer Aneinanderlagerung der Ketten, wenn die Abfolge der CH3-Seitengruppen mit der Nachbarkette übereinstimmt. Eine Kristallisation wird dadurch deutlich erschwert oder sogar verhindert. Ataktische Polymere kristallisieren nur, wenn die Seitengruppen (Substituenten) sehr klein sind, wie beim Polyvinylfluorid.
Ähnliche Probleme ergeben sich bei der dichten parallelen Anordnung der Ketten, wenn größere Seitengruppen vorhanden sind. Prinzipiell gilt: je größer die Seitengruppen werden, umso schlechter kristallisiert das Polymer. Duroplaste oder Elastomere können sich aufgrund der Vernetzung der Ketten nicht kristallin anordnen. Auch bei stark verzweigten Polymeren wie Silikonen ist eine parallele Anordnung der Ketten ausgeschlossen.
Grundsätzlich ist die Konformation der Makromoleküle im Kristall durch zwei Strukturen bestimmt: Beispielsweise liegen in Polyethylen, Polyestern und Polyamiden die Moleküle entsprechend der Bindungswinkel in Zick-Zack-Form vor. In Polyoxymethylen, Polypropylen und isotaktischen Polystyrol haben die Moleküle eine schraubenförmige (helicale) Anordnung. Die Moleküle werden dabei durch Zwischenmolekulare Kräfte stabilisiert, die im Falle der helicalen Anordnung auch intramolekular wirken.
Keimbildung
Die ersten Kristallite bilden sich z. B. infolge der Wärmebewegungen der Moleküle, wobei sich Ketten oder Kettenabschnitte in günstigen Positionen zueinander befinden und sich parallel aneinanderlagern (thermische oder homogene Keimbildung). Ein weiteres Wachstum ist jedoch aus thermodynamischen Gründen nur dann möglich, wenn Keime entstehen, die eine kritische minimale Größe überschritten haben. Ansonsten zerfallen die gebildeten Keime aufgrund thermodynamischer Instabilität wieder.[4]
Häufiger als die thermische Keimbildung ist in realen Schmelzen jedoch die Keimbildung aufgrund von Verunreinigungen oder nicht aufgeschmolzenen Kristallen. Diese wird auch als heterogene Keimbildung bezeichnet.[4] Verarbeitungshilfsmittel, Farbstoffe, Füllstoffe oder natürlich speziell zugegebene Nukleierungsmittel (Keimbildner) können die Bildung von Keimen extrem vorantreiben. Obwohl es viele Arbeiten zum Thema Nukleierungsmittel gibt, ist deren Effektivität weitgehend unverstanden. Nukleierungsmittel, die für eine Polymerart einen großen Einfluss zeigen, bleiben bei anderen Polymerarten wirkungslos.[4] Viele der bisher bekannten guten Nukleierungsmittel sind Metallsalze organischer Säuren, die bei den Kristallisationstemperaturen des Polymers bereits in kristalliner Form vorliegen.
Kristallwachstum
.svg.png.webp)
Kristallwachstum geschieht durch gefaltete Anlagerung weiterer Polymerkettenabschnitte (siehe vorangegangener Abschnitt Kristallitbildung). Dieses geschieht in einem Temperaturbereich tief genug unterhalb der Schmelztemperatur Tm und oberhalb der Glasübergangstemperatur Tg. Bei zu hoher Temperatur würden die angelagerten Ketten durch thermische Bewegungen wieder abgelöst. Unterhalb der Glasübergangstemperatur ist die Mobilität der Ketten zu gering und die Bewegung der Molekülketten ist eingefroren.[5]
Zwischen den einzelnen, parallel angeordneten Kettenabschnitten wirken zwischenmolekulare Kräfte. Je nach Art der beteiligten Atomarten können dieses Dipolwechselwirkungen oder auch Wasserstoffbrückenbindungen sein. Die Größe der Kräfte hängt außer von der Art der Wechselwirkung auch vom Abstand der parallelen Kettenabschnitte ab und bestimmt die mechanischen und thermischen Eigenschaften des Polymers.[6]
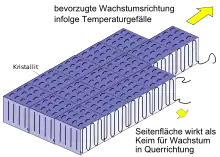
Das Wachstum der kristallinen Bereiche erfolgt bevorzugt in Richtung des größten Temperaturgradienten (Abb. 3). Die Seitenflächen wirken zwar ebenfalls als Keim für die Kristallisation; die Wachstumsgeschwindigkeit ist hier jedoch deutlich geringer. An der Ober- und Unterseite der Kristallite befinden sich die amorph wirkenden Faltungsbögen, so dass in dieser Richtung kein Wachstum stattfinden kann. Durch das gerichtete Wachstum entstehen lange, lamellenartige Bänder mit hoher Kristallinität, die ausgehend vom Kristallisationskeim wachsen und als Lamellen bezeichnet werden (Abb. 4).[4]
Die Lamellen bilden den Grundbaustein weiterer, größerer kristalliner Überstrukturen. Bei weitgehend isotropen, statischen Abkühlbedingungen bilden sich daraus Sphärolithe (Abb. 4), die aus radialsymmetrisch angeordneten Lamellen bestehen und im Hauptartikel Sphärolith eingehender beschrieben sind.
Liegt hingegen ein starkes Temperaturgefälle in der Probe vor, so kommt es zu einer weitgehend parallelen Anordnung der Lamellen und damit zu einer gerichteten, als dendritisch bezeichneten Überstruktur.[7] Derartige Strukturen werden z. B. bei Polypropylen in den oberflächennahen Randzonen beobachtet, wenn die Formtemperatur beim Spritzguss relativ kalt gewählt wird.
Bei langsam fließenden Polymeren bilden sich bei der Abkühlung hantelförmige Strukturen aus, die in der Literatur auch als sogenannte Shish-Kebab-Strukturen beschrieben werden.[2] Der Innere Teil (Seele) besteht aus parallel angeordneten, weitgehend gestreckten Ketten, während die Hanteln aus gefalteten Lamellen aufgebaut sind.
Bauteile, die sehr schnell abgekühlt wurden (niedrige Formtemperatur), hatten zu wenig Zeit, um vollständig auszukristallisieren. Hier kann es zu einem späteren Zeitpunkt (teilweise auch über Jahre) zu einer Nachkristallisation kommen. Bei diesem sekundären Kristallwachstum ändern sich die mechanischen Bauteileigenschaften. Da die Ketten im kristallinen Zustand dichter gepackt sind, kommt es außerdem zu einer Nachschwindung, d. h. zu einer nachträglichen Volumenabnahme. Dieses muss beim Spritzgussprozess berücksichtigt werden.[8]
Zum Teil werden Polymere zusätzlich längere Zeit knapp unterhalb des Schmelzpunktes gelagert, um so die Kristallinität zu erhöhen. Dieser als Tempern bezeichnete Prozess erlaubt eine höhere Ausrichtung der Polymerketten und verhindert außerdem die anschließende Nachschwindung im Einsatz.
Kristallisation durch Verstreckung
Die Kristallisation, wie oben (Kristallitbildung beim Erstarren aus der Schmelze) beschrieben, ist besonders wichtig beim Spritzgießen von Kunststoffbauteilen. Das Polymer kann während der Abkühlung meist als ruhende, relaxierte Schmelze angesehen werden.
.svg.png.webp)
Andere Bedingungen ergeben sich bei der Extrusion. Dieses Verfahren wird z. B. bei der Herstellung von Chemiefasern und Kunststofffolien eingesetzt. Das Polymer wird durch eine Düse gepresst und die Molekülketten dabei leicht vororientiert.
Durch eine Nachverstreckung (Anlegen einer Zugspannung) kann die Orientierung der Molekülketten deutlich erhöht werden. Fasern werden z. B. auf ein Vielfaches ihrer ursprünglichen Länge gezogen. Dabei werden die Ketten gereckt und orientiert angeordnet. Dieser Zustand entspricht einer Teilkristallisation, wobei die kristallinen Bereiche zusätzlich noch gleichgerichtet sind.[6] Die Festigkeit der Faser in Längsrichtung wird stark erhöht. Normalerweise erfolgt eine anschließende Temperaturbehandlung unter Spannung (Thermofixieren), um eine höhere Ordnung zu erreichen und Spannungen abzubauen, die zu einer nachträglichen Relaxation (Schrumpf) führen könnten.[2][9] Die Faser bleibt dadurch formstabiler. Die starke Anisotropie der Faser wird auch in ihren optischen Eigenschaften (Doppelbrechung) messbar.
Eine Festigkeitssteigerung durch nachträgliche Verstreckung wird auch beim Prozess des Streck-Blasformens erzeugt. Hier wird der temperierte PET-Rohling (engl. Preform) in einem Umformprozess mit Druckluft bis zur durch die Form vorgegebenen Größe aufgeblasen. Anwendungen sind z. B. Benzintanks oder PET-Flaschen.[6] Gleichzeitig kann die Gasdurchlässigkeit durch die biaxiale (in zwei Richtungen weisende) Verstreckung deutlich verringert werden.[10]
Durch eine nachträgliche Verstreckung können auch an sich amorphe Polymere zu teilkristallinen Materialien umgeformt werden.[10] Es bilden sich lamellare Kristallstrukturen aus, die infolge der starken Verstreckung keine sphärolithischen Überstrukturen bilden und damit optisch vollständig transparent bleiben.[10]
Kristallisation aus der Lösung
Polymere können auch aus einer Lösung oder durch Verdampfung eines Lösungsmittels kristallisiert werden. Lösungen unterscheidet man nach dem Verdünnungsgrad. In verdünnten Lösungen haben die Molekülketten keine Verbindung untereinander und liegen als separate Polymerknäuel in der Lösung vor. Wird die Konzentration des Polymers in der Lösung erhöht (konzentrierte Lösung), so durchdringen sich die Ketten gegenseitig immer mehr, und es kommt bei weiterer Reduktion des Lösungsmittels (z. B. durch Verdampfen) zu einer Ordnung der Polymerketten zu Kristalliten.[11] Der Vorgang entspricht weitgehend der Kristallisation aus der Schmelze.
Mit Hilfe der hochauflösenden magnetischen Kernspinresonanz wird nur der gelöste Anteil einer übersättigten Polymer-Lösung erfasst. Damit kann die Abnahme des gelösten Anteils während der Kristallisation aus der Lösung und daraus die Kristallisationsgeschwindigkeit bestimmt werden.[11]
Eine spezielle Form der Kristallisation kann beobachtet werden, wenn einige Milliliter einer heißen Lösung von Polyethylen in Xylol (90 °C) auf die Oberfläche von Wasser mit der gleichen Temperatur gegossen werden. Es entsteht bei der Verdunstung des Lösungsmittels eine dünne Haut (etwa 1 Mikrometer dick) mit einem wabenartigen Aufbau. Bei der Betrachtung im Polarisationsmikroskop erkennt man bei gekreuzten Polarisationsfiltern, dass es sich um Sphärolithe mit negativer Doppelbrechung in radialer Richtung handelt. Das bedeutet: Der Brechungsindex ist für in radialer Richtung schwingendes Licht kleiner als für die tangentiale Schwingungsrichtung. Außer dem für Sphärolithe charakteristischen Auslöschungskreuz beobachtet man periodische dunkle Ringe als geometrische Orte für Stellen, an denen der Beobachter in Richtung der optischen Achse blickt. Die Lamellen, aus denen die Sphärolithe bestehen, sind schraubenartig um den Radius verdreht. Beim Zerreißen der Haut kommt es zur faktisch übergangslosen Umwandlung der Lamellen in Fasern mit positiver Doppelbrechung.[12]
 Haut aus Polyethylen, kristallisiert bei 90 Grad Celsius aus einer Lösung in Xylol auf einer Wasseroberfläche
Haut aus Polyethylen, kristallisiert bei 90 Grad Celsius aus einer Lösung in Xylol auf einer Wasseroberfläche Spärolithe in einer Haut aus Polyethylen, Polarisationsmikroskop
Spärolithe in einer Haut aus Polyethylen, Polarisationsmikroskop Umwandlung von Lamellen in Fasern beim Zerreißen einer Haut aus Polyethylen
Umwandlung von Lamellen in Fasern beim Zerreißen einer Haut aus Polyethylen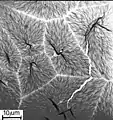 Haut aus Polyethylen, kristallisiert bei 90 Grad Celsius aus einer Lösung in Xylol auf einer Wasseroberfläche, Foto mit Elektronenmikroskop
Haut aus Polyethylen, kristallisiert bei 90 Grad Celsius aus einer Lösung in Xylol auf einer Wasseroberfläche, Foto mit Elektronenmikroskop
Kristallinität, Kristallinitätsgrad, Kristallisationsgrad
| Polymerart | typischer Kristallisationsgrad[2] |
|---|---|
| Polyamid (PA66 und PA6) | 35…45 % |
| Polyoxymethylen (POM-Homopolymer) | 90 % |
| Polyoxymethylen (POM-Copolymer) | 75 % |
| Polyethylenterephthalat (PET) | 30…40 % |
| Polybutylenterephthalat (PBT) | 40…50 % |
| Polytetrafluorethylen (PTFE) | 60…80 % |
| Polypropylen (PP), isotaktisch | 70…80 % |
| Polypropylen (PP), syndiotaktisch | ≈ 30…40 % |
| Polypropylen (PP), ataktisch | ≈ 0 % |
| Polyethylen hoher Dichte (PE-HD) | 70…80 % |
| Polyethylen niedriger Dichte (PE-LD) | 45…55 % |
Die Begriffe Kristallinität, Kristallinitätsgrad und Kristallisationsgrad werden in der Literatur als Synonyme verwendet und bezeichnen jenen Anteil eines teilkristallinen Feststoffes, der kristallin ist. Bei Polymeren hängt der Kristallisationsgrad von der thermischen Vergangenheit des Materials ab.
Typischerweise werden technisch Kristallisationsgrade von 10 bis 80 % realisiert.[2] Das Erreichen von höheren Kristallinitäten ist nur möglich bei niedermolekularen Materialien und/oder speziell getemperten Proben. Im ersten Fall wird das Material dadurch spröde, im letzten Fall bedeutet die lange Lagerung bei Temperaturen knapp unter dem Schmelzpunkt (Tempern) deutliche Kosten, welche sich nur in Spezialfällen rechnen. Kristallinitäten unter 10 % führen zu einer zu hohen Kriechneigung, wenn die Anwendungstemperatur des Bauteils oberhalb der Glasübergangstemperatur Tg liegt.
Der Kristallisationsgrad wird in der Regel als Massenbruch oder Molenbruch angegeben. Vereinzelt gibt es auch noch die Angabe einer volumenbezogenen Angabe des Kristallisationsgrades.
Die meisten Auswertungen von Kennzahlen des Kristallisationsgrades für teilkristalline Thermoplasten gehen von einem Zweiphasenmodell aus, bei dem es perfekte Kristalle und eindeutige amorphe Bereiche gibt. Die Abweichungen durch Fehlstellen, Übergangsbereiche zwischen amorph und kristallin dürften bis zu einigen Prozent betragen.[2]
Die gängigsten Methoden zur Bestimmung des Kristallisationsgrades bei Polymeren sind die Dichtemessung, DSC, Röntgenbeugung, IR-Spektroskopie oder NMR. Der ermittelte Messwert hängt von der verwendeten Messmethode ab.[13] Deshalb sollte zusätzlich zum Kristallisationsgrad immer die Methode mit angegeben werden.[2]
Neben den oben genannten integralen Methoden kann die Verteilung von kristallinen und amorphen Bereichen über mikroskopische Verfahren (speziell Polarisationsmikroskopie und Transmissionselektronenmikroskopie) visualisiert werden.
- Dichtemessungen
- Kristalline Bereiche sind im Allgemeinen dichter gepackt als amorphe Bereiche. Daraus resultiert eine höhere Dichte, die sich je nach Material typischerweise um bis zu ca. 15 % unterscheidet (Beispiel Polyamide 6: und ). Die kristalline Dichte wird dabei aus dem kristallinen Aufbau berechnet, während die amorphe Dichte experimentell an abgeschrecktem, amorphem Material gemessen wird. Das Problem der Dichtemessung zur Bestimmung der Dichte-Kristallinität ist, dass die Dichte der amorphen Bereiche von den Abkühlbedingungen abhängig ist und in der Probe vorhandene Feuchte den Messwert beeinflussen kann.[5]




- Kalorimetrie (DSC)
- Beim Schmelzen teilkristalliner Polymere muss zur Umwandlung der festen kristallinen Strukturen in einen amorphen flüssigen Zustand zusätzliche Energie aufgewendet werden. Der Analytiker spricht hier von einer endothermen Enthalpieänderung. Der Vorgang erstreckt sich über einen größeren Temperaturbereich. Zunächst schmelzen die kleineren oder weniger regelmäßig aufgebauten Kristalle. Mit zunehmender Temperatur schmelzen dann immer dickere und größere Kristallite, bis die gesamte Probe aufgeschmolzen ist.[14]
Die Schmelzenthalpie (notwendige Energie zum Aufschmelzen der Kristalle) kann mit Hilfe der Dynamische Differenzkalorimetrie (DSC) gemessen werden. Durch den Vergleich mit einem Literaturwert für vollständig kristallines Material (Kristallisationsgrad von 100 %) kann der kalorimetrische Kristallisationsgrad der Probe berechnet werden.[5]
- Röntgenbeugung
- Immer wiederkehrende Atomabstände erzeugen Signale bei entsprechenden Winkeln im Diffraktogramm. In amorphen Substanzen sind sehr unterschiedliche Abstände zwischen den Molekülketten vorhanden. Dieses führt zu einer sehr breiten Verteilung im Diagramm in Form einer sehr breiten Glockenkurve (Halo). Die regelmäßigen Anordnungen in kristallinen Bereichen erzeugen hingegen sehr viel schmalere Verteilungen in Form von Peaks. In Diffraktogrammen realer Polymere sind Halo als auch Peaks überlagert. Durch eine Peakentfaltung können die Intensitäten der Peaks und des Halos ermittelt und daraus die Röntgen-Kristallinität berechnet werden.[5]
- Infrarotspektroskopie (IR)
- In den IR-Spektren findet man bei kristallinen Polymeren zusätzliche Signale (Banden), die bei amorphen Materialien gleicher Zusammensetzung fehlen. Diese Banden stammen von Deformationsschwingungen die durch die regelmäßige Anordnung der Molekülketten erst ermöglicht werden. Aus der Auswertung dieser Banden kann der Infrarot-Kristallisationsgrad berechnet werden.[5]
- Kernresonanzspektroskopie (NMR)
- Kristalline und amorphe Bereiche unterscheiden sich in der Protonenbeweglichkeit. Dieses zeigt Auswirkungen in der Linienform im Spektrum. Unter Berücksichtigung des Strukturmodells können hieraus Aussagen über die Kristallinität getroffen werden.[5]
Eigenschaften teilkristalliner Polymere
| Eigenschaften nehmen zu | Eigenschaften nehmen ab |
|---|---|
| Steifigkeit, Modul | Schlagzähigkeit |
| Dichte | Dehnung |
| Streckspannung | Thermische Ausdehnung |
| Chemikalienbeständigkeit | Permeabilität |
| Glas- und Schmelztemperatur | Quellungsverhalten |
| Abrasionswiderstand | Mechanische Dämpfung |
| Dimensionsstabilität | Kriechneigung |
Das technische Verhalten und die Eigenschaften von Kunststoffen werden durch die chemische Natur der Grundbausteine, der Länge, aber auch der Anordnung der Makromoleküle bestimmt.[15]
Die Kristallisation der Makromoleküle verändert die Eigenschaften eines Materials erheblich. Die Eigenschaften eines teilkristallinen Werkstoffes werden sowohl von den kristallinen als auch von den amorphen Bereichen des Polymers bestimmt. Dadurch ist ein gewisser Zusammenhang mit Kompositmaterialien zu sehen, die ebenfalls aus mehreren Substanzen aufgebaut sind. Typische Eigenschaftsänderungen bei Zunahme der Kristallisation sind in der nebenstehenden Tabelle zusammengefasst und werden im Folgenden genauer beschrieben.
Thermische Eigenschaften
Unterhalb ihrer Glasübergangstemperatur besitzen amorphe Polymerbereiche spröde, hartelastische Eigenschaften. Dieses ist auf die Unbeweglichkeit der eingefrorenen Ketten zurückzuführen. Wird die Glasübergangstemperatur (auch Erweichungstemperatur genannt) überschritten, so werden die Molekülketten gegeneinander beweglich, und es entstehen die typischen gummielastischen Eigenschaften des Kunststoffs. Mit zunehmender Temperatur wird die Beweglichkeit der Ketten immer höher, und das Material somit immer weicher. Der Elastizitätsmodul nimmt deutlich ab. Bei ständiger Krafteinwirkung auf das Bauteil kommt es zu einer viskoelastischen Verformung, d. h. das Polymer beginnt zu kriechen. Eine Wärmeformbeständigkeit ist somit für amorphe Polymere nur unterhalb der Glasübergangstemperatur gegeben.[16]
Zwischen den Ketten der kristallin angeordneten Bereiche wirken zwischenmolekulare Kräfte, welche die Erweichung verhindern. Oberhalb der Glasübergangstemperatur ist der Elastizitätsmodul immer noch relativ hoch. Die Kristallite schmelzen erst bei einer wesentlich höheren Schmelztemperatur unter Zufuhr deutlicher Energiemengen, welche nötig sind um die regelmäßige Anordnung der Ketten zu überwinden (Schmelzenthalpie). Erst bei diesem Übergang zur viskosen Schmelze erfolgt ein starker Abfall des Elastizitätsmoduls. Teilkristalline Polymere sind damit bei wesentlich höheren Temperaturen einsetzbar, ohne dass das entsprechende Bauteil seine Dimension oder Gestalt verändert.[2]
Bei abgeschreckten (nicht auskondensierten) Materialien kann es zu einer Nachkondensation kommen, die eine Schwindung des Bauteils bewirkt (vgl. Abschnitt Kristallwachstum).
Mechanische Eigenschaften
Die mechanischen Eigenschaften des Polymers setzen sich aus den Eigenschaften der kristallinen und der amorphen Bereiche zusammen. Je höher der Anteil dicht gepackter Kristallite, desto härter, allerdings auch spröder, wird das Bauteil. Für die Herstellung von Kunststoffgegenständen ist also eine gewisse Kristallinität durchaus erwünscht, da diese für die Stabilität des Kunststoffes verantwortlich ist. Die amorphen Bereiche sind hingegen nötig, um den makromolekularen Werkstoffen eine gewisse Elastizität und Schlagzähigkeit zu geben.[4]
Kunststoffe sind viskoelastische Stoffe, was bedeutet, dass das Werkstoffverhalten bei äußerer Beanspruchung eine Funktion der Zeit ist. Bei konstanter Last nimmt die Deformation mit der Zeit zu (Kriechen, Retardieren). Bei konstanter Verformung nimmt die Spannung mit der Zeit ab (Erholen, Relaxieren). Zur Beschreibung der mechanischen Eigenschaften werden daher normalerweise Spannungs-Dehnungs-Diagramme aufgenommen. Man unterscheidet das Kurzzeitverhalten (z. B. Zugversuch mit typischen Zeiten im Minutenbereich), die stoßartige Beanspruchung, das Verhalten bei langzeitiger und ruhender Beanspruchung, wie auch die schwingende Beanspruchung.[15]
Bei der plastischen Verformung von teilkristallinen Polymeren geht man von einer Orientierung der gefalteten Kristallite und einem ‚Abspulen‘ bzw. Abgleiten vorher gefalteter Molekülketten aus. Durch das Abspulen der Ketten kommt es im Bereich der Deformationszone zu einer plastischen Verformung in Form einer starken Einschnürung (engl. Neck für Hals) bei gleichzeitiger Ausrichtung der Molekülketten in Zugrichtung.[1] Verstreckte Materialien und damit ausgerichtete Molekülketten können nur noch sehr wenig gedehnt werden. Diesen Effekt macht man sich z. B. bei synthetischen Fasern zu Nutze. Die zahlreichen in Zugrichtung verlaufenden Molekülketten verstärken sich gegenseitig und sorgen für eine deutliche erhöhte Festigkeit in Faserrichtung.
Auch die Molekülmasse (Kettenlänge) hat einen Einfluss auf die Polymereigenschaften. Mit steigender Kettenlänge erhöhen sich die Berührungsflächen, was zu einer Erhöhung der Zugfestigkeit und einer Erhöhung der chemischen Beständigkeit führt. Gleichzeitig nimmt die Zahl der Verschlaufungen zu, was die Zähigkeit bei Raumtemperatur verbessert, aber auch das Fließverhalten der Schmelze negativ beeinflusst. Wenn die zwischenmolekularen Kräfte stärker werden als die Kettenfestigkeit, so nimmt die Zugfestigkeit trotz steigender Kettenlänge nicht mehr zu.[6]
Dichte und Permeabilität
.svg.png.webp)
Wird einer Kunststoffschmelze Wärme entzogen, so verringert sich die Beweglichkeit der Ketten. Das Volumen reduziert sich bei amorphen Materialien zuerst weitgehend linear mit der Temperatur. Unterhalb der Glasübergangstemperatur Tg sind die Ketten unbeweglich. Der Wärmeausdehnungskoeffizient ändert sich hier, woraus die abweichende Steigung der roten Kurve in Abbildung 7 resultiert.
Bei kristallinen Materialien kommt es unterhalb der Schmelztemperatur Tm zu einer regelmäßigen Anordnung der Molekülketten (Ordnungszustand) und damit zu einer deutlichen Verringerung des Abstands zwischen den Ketten durch zwischenmolekulare Kräfte. Dieses führt zu einer Erhöhung der Dichte bzw. Reduzierung des spezifischen Volumens (hellblaue Kurve in Abbildung 7).[17]
Durch die dichtere Packung der Ketten kann Gas schlechter durchgelassen werden, was zu einer Verringerung der Permeabilität, oder anders ausgedrückt, zu einer Erhöhung der Gasdichtheit führt.
Optische Eigenschaften
In der Regel sind teilkristalline Polymere opak, d. h. eingetrübt. Das liegt an der Lichtbrechung aufgrund der unterschiedlichen Brechungsindices von kristallinen und amorphen Bereichen. Der Eintrübungsgrad nimmt mit der Kristallinität zu[4], hängt aber auch von Unterschieden im Brechungsindex ab. So ist z. B. syndiotaktisches Polypropylen fast vollständig durchsichtig, während isotaktisches Polypropylen mit vergleichbarer Kristallinität von ca. 50 % stark opak ist. Das lässt sich durch die unterschiedliche Kristallstruktur dieser beiden Modifikationen erklären.
Die nachträgliche Anfärbung erfolgt maßgeblich über die amorphe Phase. Die Farbstoffmoleküle können besser zwischen die Molekülketten des Polymers dringen. Materialien mit höherem Kristallisationsgrad lassen sich daher schlechter Anfärben als Materialien mit mehr amorphen Bereichen aber ansonsten gleicher Zusammensetzung.[9]
Einfluss der Kristallisation auf die Verarbeitungseigenschaften beim Spritzguss
Beim Spritzgießen teilkristalliner Thermoplaste muss beachtet werden, dass die durch den Kristallisationsvorgang zusätzlich freiwerdende Wärme abgeführt wird, wodurch sich die Zykluszeit verlängert. Zusätzlich muss die größere Volumenänderung des Materials (aufgrund der Dichteänderung bei der Kristallisation) durch längere Nachdruckzeiten ausgeglichen werden.[17]
Bei teilkristallinen Thermoplasten ist außerdem die Schwindung größer als bei amorphen Materialien. Die Abkühlbedingungen müssen genau eingehalten werden, da der Abkühlvorgang den Kristallisationsgrad und damit die Material- und Formteileigenschaften nachhaltig beeinflusst. Sehr schnelles Abkühlen gestattet es zwar die Kristallisation weitgehend zu unterdrücken und eine nahezu amorphe Erstarrung zu erzwingen, allerdings kommt es in diesem Fall mit der Zeit zu einer Nachkristallisation, was eine weitere Schwindung und Verzug bedeutet.[17]
Geschichte und weitere Kristallisationsmodelle
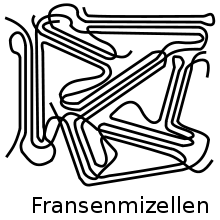
1925 fand Hermann Staudinger heraus, dass gewisse chemische Substanzen aus langkettigen Molekülen bestehen. Röntgenstrukturuntersuchungen zeigten (je nach Material) Beugungsspektren, wie sie für Kristalle typisch sind. Genauere Untersuchungen ergaben, dass manche Polymere aus vielen kleinen, kristallinen Strukturen aufgebaut sein müssen. Mit den aus den röntgenografischen Untersuchungen erhaltenen ‚Gitterkonstanten‘ und der bekannten chemischen Zusammensetzung berechnete man die Dichte des Materials. Die berechnete Dichte war jedoch immer höher als die experimentell bestimmte Dichte des Polymers.[1]
Daraufhin wurde von Abitz und Gerngroß das Modell der Fransenmizelle entwickelt (Abb. 6).[8] Dabei seien Abschnitte der Molekülketten parallel zueinander als Kristall angeordnet. Die Zwischenbereiche seien hingegen amorph. Eine Molekülkette (so die Vorstellung) durchlaufe dabei verschiedene Kristalle. Nachdem 1957 erstmals kleinste polymere Einkristalle hergestellt wurden, zeigte sich, dass das Modell der Fransenmizelle für die Beschreibung von Einkristallen nicht aufrechterhalten werden konnte.[1] A. Keller postulierte 1957 in der Zeitschrift Nature den Aufbau von Kristalliten in Form der oben beschriebenen gefalteten Molekülketten (Abb. 3), die von einer Seite der Lamelle zur anderen Seite und wieder zurück verlaufen.[18]
G. Kanig hat mit der von ihm 1975 entwickelten Kontrastiermethode nicht nur die Lamellenstruktur von Polyethylen elektronenmikroskopisch sichtbar gemacht, sondern konnte auch ihre Entstehung beim Abkühlen aus der Schmelze, bzw. ihr Aufschmelzen bei Erwärmung des Materials beobachten.[18]
Siehe auch
Literatur
- Bernd Tieke: Makromolekulare Chemie. Eine Einführung. Wiley-VCH, Weinheim 2000. ISBN 978-3-527-29364-3.
- Georg Menges, Edmund Haberstroh, Walter Michaeli, Ernst Schmachtenberg: Werkstoffkunde Kunststoffe. Hanser Verlag, 2002, ISBN 3-446-21257-4 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Wolfgang Weißbach: Werkstoffkunde und Werkstoffprüfung. Vieweg+Teubner Verlag, 2007, ISBN 3-8348-0295-6 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Friedrich Johannaber, Walter Michaeli: Handbuch Spritzgießen. Hanser Verlag, 2004, ISBN 3-446-22966-3 (Eingeschränkte Vorschau in der Google-Buchsuche).
- G.W. Becker, Ludwig Bottenbruch, Rudolf Binsack, D. Braun: Technische Thermoplaste. 4. Polyamide. Hanser Verlag, 1998, ISBN 3-446-16486-3 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Wilbrand Woebcken, Klaus Stoeckhert, H. B. P. Gupta: Kunststoff-Lexikon. Hanser Verlag, 1998, ISBN 3-446-17969-0 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Michael Dröscher: Ordnungszustände in Polymeren. In: Chemie in unserer Zeit. Band 10, Nr. 4, 1976, S. 106–113, doi:10.1002/ciuz.19760100403.
Einzelnachweise
- Martin Bonnet: Kunststoffe in der Ingenieuranwendung: Eigenschaften, Verarbeitung und Praxiseinsatz polymerer Werkstoffe. Vieweg+Teubner Verlag, 2008, ISBN 3-8348-0349-9 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Gottfried W. Ehrenstein: Polymer-Werkstoffe. Hanser Fachbuch, 1999, ISBN 3-446-21161-6 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Andrew Keller: Crystalline Polymers; an Introduction. In: Faraday Discussion of the Royal Society of Chemistry 1979. Band 68. Faraday Division of the Royal Society of Chemistry, London 1979, S. 149 (englisch).
- Georg Menges, Edmund Haberstroh, Walter Michaeli, Ernst Schmachtenberg: Werkstoffkunde Kunststoffe. Hanser Verlag, 2002, ISBN 3-446-21257-4 (Eingeschränkte Vorschau in der Google-Buchsuche).
- G.W. Becker, Ludwig Bottenbruch, Rudolf Binsack, D. Braun: Technische Thermoplaste. 4. Polyamide. Hanser Verlag, 1998, ISBN 3-446-16486-3 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Wolfgang Weißbach: Werkstoffkunde und Werkstoffprüfung. Vieweg+Teubner Verlag, 2007, ISBN 3-8348-0295-6 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Institut für den Wissenschaftlichen Film, Klaus Peter Großkurth: Kristallisation von Polypropylen, 1990, doi:10.3203/IWF/C-1699 (Video mit Erklärungen zur dendritischen Kristallisation von Polypropylen).
- Wilbrand Woebcken, Klaus Stoeckhert, H. B. P. Gupta: Kunststoff-Lexikon. Hanser Verlag, 1998, ISBN 3-446-17969-0 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Burkhard Wulfhorst: Textile Fertigungsverfahren. Hanser Verlag, 1998, ISBN 3-446-19187-9 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Michael Thielen, Klaus Hartwig, Peter Gust: Blasformen von Kunststoffhohlkörpern. Hanser Verlag, 2006, ISBN 3-446-22671-0, (Eingeschränkte Vorschau in der Google-Buchsuche).
- J. Lehmann: Die Beobachtung der Kristallisation hochpolymerer Substanzen aus der Lösung durch Kernspinresonanz. In: Colloid & Polymer Science. Band 212, Nr. 2, 1966, S. 167–168, doi:10.1007/BF01553085.
- Heinz H. W. Preuß: Bruchflächenmorphologie und Charakter des Bruches von Polyäthylenkörpern, Dissertation, Leipzig, 1963.
- M. D. Lechner, K. Gehrke und E. H. Nordmeier: Makromolekulare Chemie, 4. Auflage, Birkhäuser Verlag, S. 355, ISBN 978-3-7643-8890-4.
- Gottfried W. Ehrenstein, Gabriela Riedel, Pia Trawiel: Praxis der thermischen Analyse von Kunststoffen. Hanser Verlag, 2003, ISBN 3-446-22340-1 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Walter Michaeli: Einführung in die Kunststoffverarbeitung. Hanser Verlag, 2006, ISBN 3-446-40580-1 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Joachim Nentwig: Kunststofffolien. Hanser Verlag, 2006, ISBN 3-446-40390-6, (Eingeschränkte Vorschau in der Google-Buchsuche).
- Friedrich Johannaber, Walter Michaeli: Handbuch Spritzgießen. Hanser Verlag, 2004, ISBN 3-446-22966-3 (Eingeschränkte Vorschau in der Google-Buchsuche).
- Hans K. Felger, Hermann Amrehn, Alexander von Bassewitz, Gerhard W. Becker: Polyvinylchlorid. Hanser Verlag, 1986, ISBN 3-446-14416-1 (Eingeschränkte Vorschau in der Google-Buchsuche).
Weblinks
- Video: Kristallisation von Polypropylen. Institut für den Wissenschaftlichen Film (IWF) 1988, zur Verfügung gestellt von der Technischen Informationsbibliothek (TIB), doi:10.3203/IWF/C-1699.