Intrinsisch ungeordnete Proteine
Ein intrinsisch ungeordnetes Protein (Kurzbezeichnung: IDP, englisch: intrinsically disordered protein) ist ein Protein, dem eine feste oder geordnete dreidimensionale Struktur fehlt.[2][3][4] IDPs umfassen vollständig unstrukturierte und teilstrukturierte Proteine, die unter anderem Random Coils und (pre-)molten globules enthalten. Dazu zählen auch Proteine mit mehrteiligen Domänen, dessen Domänen mit kurzen Peptidsequenzen (flexible linkers) verbunden sind. In einigen Fällen können IDPs durch das Binden an andere Makromoleküle in eine feste Struktur überführt werden.[5]
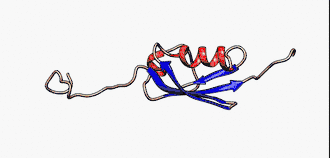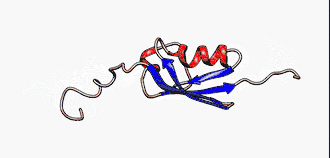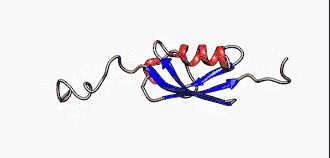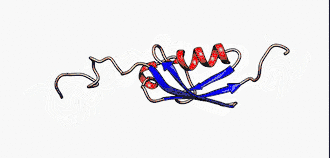
Sie sind nicht zu verwechseln mit den intrinsisch unstrukturierten Proteinen, die eine Untergruppe der intrinsisch ungeordneten Proteinen darstellt. Jedoch werden die Begriffe in der Literatur manchmal synonym verwendet.
Geschichte
Das Anfinsen-Dogma besagt, dass die native Struktur von Proteinen unter physiologischen Bedingungen durch die Aminosäuresequenz bestimmt wird. In den 1960er-Jahren konnte man zeigen, dass bei Denaturierung von Ribonukleasen 99 % ihrer enzymatischen Aktivität verloren ging und bei Wiedereinführung in ihre physiologische Bedingung ihre enzymatische Aktivität zurückerhalten hatten.[6] Daraus konnte man schlussfolgern, dass allein die Aminosäuresequenz ausreichte, um das Protein in eine funktionale Konformation mit niedrigster freier Energie zu falten. 1972 erhielten Christian B. Anfinsen, Stanford Moore und William Howard Stein für diese Entdeckung den Nobelpreis für Chemie.
Anfang der 2000er-Jahre erkannte man, dass nicht alle Proteine im gefalteten Zustand funktionierten.[7] Daher müssten einige Proteine ungefaltet oder ungeordnet vorliegen, um ihre Funktion auszuüben.[8] Schätzungen zufolge sind ca. 10 % der Proteine ungeordnet und 40 % der eukaryotischen Proteine haben zumindest eine lange (> 50 Aminosäuren) ungeordnete β-Schleife.[9] Die Aminosäuresequenzen zeigten unter physiologischen Bedingungen in vitro ähnliche physikochemische Merkmale wie die von Random Coils auf. Random Coils sind ebenfalls wenig bis gar nicht strukturiert, besitzen keinen fest gepackten Kern, aber haben dafür eine erweiterte Konformation mit hoher intramolekularer Flexibilität.
Bei vielen kristallographischen Strukturen fehlten β-Schleifen und dies wurde anhand einer Reihe von Aminosäuren mit fehlenden Atomkoordinaten im Modell ersichtlich. Man dachte, dass die „Lücken“ im Modell Artefakte von zufälligen Störungen im Kristall waren. In einigen Fällen können diese Lücken auf intrinsisch ungeordnete β-Schleifen in einem sonst gefalteten Protein hinweisen.[10] Solche Lücken sind Grundlagen für Server wie DISOPRED3, das intrinsisch ungeordnete Regionen (IDR, engl. intrinsically disordered region) und Proteinbindungsstellen innerhalb dieser Regionen vorhersagen kann.
Im Jahr 2011 wurde von Tanguy Chouard in der Fachzeitschrift Nature ein umfassender Überblick und einige Funktionen von IDPs veröffentlicht.[11]
Eigenschaften
Die Regulierung durch posttranslationale Modifikation führt zur erhöhten Bindungsaffinität zwischen den IDPs und deren Rezeptoren. Es wurde vorgeschlagen, dass die Flexibilität von ungeordneten Proteinen die Bindung zu modifizierenden Enzymen und den jeweiligen Rezeptoren erleichtert.[12] Proteine mit intrinsischer Unordnung sind hauptsächlich in der Signaltransduktion, Transkription und der Umgestaltung des Chromatins involviert.[13][14]
Flexible linkers
Ungeordnete Regionen treten oftmals als flexible linkers oder Schleifen auf, die Proteindomänen an sich binden können. Die Aminosäuresequenzen von flexible linkers variieren stark hinsichtlich der Länge, aber sie sind reich an polaren Aminosäuren. Flexible linkers in der Lage, dass die Domänen, mit denen sie verbunden sind, sich verdrehen und rotieren können, um somit mithilfe der Proteindomänen-Dynamik Bindungspartner anziehen können. Sie binden auch an Proteine, um ihnen mehr Möglichkeiten zur Konformationsänderung durch weitreichende Allosterie zu bieten.[15][2]
Lineare Motive
Lineare Motive sind kurze Abschnitte von Proteinen, die funktionale Interaktionen mit anderen Proteinen oder Biomolekülen, wie DNA, RNA, Zucker etc. vermitteln. Sie spielen meist eine wichtige Rolle in der Zellregulation, beispielsweise die Kontrolle der Zellstruktur, subzelluläre Lokalisation von verschiedenen Proteinen sowie die Regulation des Proteinumsatzes. Meistens wird durch posttranslationale Modifikation, wie z. B. durch Phosphorylierung, die Affinität für bestimmte Interaktionen modifiziert. Mithilfe der Kernspinresonanz wurden bei 80 % der IDPs sogenannte PreSMos (pre-structured motifs) erkannt, die als kurzzeitige strukturelle Sekundärelemente bei der Zielerkennung involviert sind. In einigen Fällen werden durch Bindung an einem Zielpartner die kurzzeitigen strukturellen Sekundärelemente fest und stabilisieren sich, beispielsweise werden sie zu Helices. Vermutlich bilden die PreSMos die aktiven Zentren der IDPs.[16]
Gekoppelte Faltung und Bindung
Viele ungeordnete Proteine gehen bei Bindung in einem geordneteren Zustand über, z. B. Molecular recognition features (MoRFs).[17] Das Binden und die gekoppelte Faltung geschieht nur lokal und benötigt nur einige interagierende Aminosäurereste oder eine ganze Proteindomäne. Bestimmte ungeordnete Regionen dienen als molekulare Schalter, die bestimmte biologische Rollen übernehmen und bei Bindung in einen geordneten Zustand übergehen, beispielsweise durch Bindung an kleine Moleküle, Bindung an DNA/RNA, Ioneninteraktionen etc.[18]
Die Eigenschaft von ungeordneten Proteinen zur Bindung, um somit eine Funktion auszuüben zeigt, dass die Stabilität des Proteins keine Bedingung dafür ist, eine Funktion ausüben zu können. Viele kurze funktionale Stellen, wie zum Beispiel kurze lineare Motive, liegen im ungeordneten Protein überrepräsentiert vor. Besonders zahlreich findet man ungeordnete Proteine und kurze lineare Motive in RNA-Viren, wie Henipaviren, Hepatitis-C-Virus, HIV-1 und humane Papillomviren, um somit die Bindung und Manipulation einer Vielzahl an Proteinen in der Wirtszelle zu ermöglichen und somit ihr Genom übertragen können.[19]
Unordnung im gebundenen Zustand (Fuzzy-Komplex)
Trotz Binden an andere Proteine können sich intrinsisch ungeordnete Proteine in ihrer Konformation jederzeit ändern. Diese strukturelle Unordnung kann im gebundenen Zustand statisch oder dynamisch sein. Fuzzy-Komplexe sind Proteinkomplexe, die aus IDPs bestehen und für die Ausübung ihrer biologischen Funktion ihre Konformation im gebundenen Zustand jederzeit ändern können.[20] Die Bindungsspezifität von DNA-bindenden Proteinen kann durch alternatives Spleißen beeinflusst werden, in dem die Länge von Fuzzy-Regionen variiert wird.[21]
Struktur
Intrinsisch ungeordnete Proteinen passen sich den jeweiligen Bedingungen der Zelle durch Konformationsänderung an. Die Gesamtheit aller möglichen Konformationszustände wird als structural oder conformational ensemble bezeichnet.[22] Daher ist die Struktur von IDPs stark von deren Funktion abhängig. Jedoch liegt nur ein geringer Anteil der IDPs völlig ungeordnet (im nativen Zustand) vor. IDPs besitzen bestimmte Bereiche, die intrinsisch ungeordneten Regionen (IDRs), welche hauptsächlich den Grad der Unordnung bestimmen. Aus dem Grund kann man IDPs in vollständig ungeordnet (intrinsisch unstrukturierte Proteine) und in Proteine mit IDRs unterteilen.
Ob eine Unordnung und welche Art von Unordnung vorliegt, wird von der Aminosäuresequenz bestimmt.[2] Im Allgemeinen besitzen IDPs wenige sperrige Aminosäuren (die sterische Hinderung verursachen können) und sind meistens reich an polaren und elektrisch geladenen Aminosäuren, die eine niedrige Fettlöslichkeit (niedrige Hydrophobizität) verursachen.[22] Somit ist eine gute Interaktion mit Wasser möglich. Hohe Gesamtladungen im Protein begünstigen die Unordnung, das durch elektrostatische Abstoßung durch gleich geladene Aminosäurereste verursacht wird. Somit sind IDPs nicht in der Lage sich in globuläre Proteine zu falten, da durch die elektrostatischen Abstoßungen kein hydrophober Kern gebildet werden kann. In einigen Fällen können bestimmte hydrophobe Cluster in IDRs Aussagen darüber treffen, welche Regionen sich einer gekoppelten Faltung und Bindung (durch schwache und nicht-spezifische Bindung an das Targetprotein, auch fly casting-Effekt genannt) unterziehen.[23]
Viele IDPs besitzen keinerlei reguläre Sekundärstrukturen und werden daher als flexibel bezeichnet (die an den verschiedenen Kombinationen der Ramachandran-Winkel erkennbar sind). Dabei bezeichnet Flexibilität hierbei keinen Gleichgewichtszustand, wie das bei strukturierten Proteinen der Fall ist.[24] Ebenfalls besitzen viele IDPs sogenannte low-complexity regions (LCRs), bei denen beispielsweise Sequenzen von einigen Aminosäurereste oder Aminosäuremotive überpräsentiert vorkommen. LCRs sind mögliche Indikatoren für eine Unordnung, jedoch besitzen nicht alle IDPs low-complexity regions.
Experimenteller Nachweis
Intrinsische ungeordnete Proteine können, sobald sie gereinigt sind, durch verschiedene experimentelle Methoden nachgewiesen werden. Die wichtigste Methode, um Informationen über ungeordnete Regionen im Protein zu erhalten, ist die NMR-Spektroskopie. Bei der Kristallstrukturanalyse kann der nachgewiesene Mangel an Elektronendichte ebenfalls ein Nachweis für ungeordnete Regionen sein.
Gefaltete Proteine besitzen eine hohe Dichte (mit einem partiellen spezifischen Volumen von 0,72–0,74 mL/g) und einen kleinen Streumassenradius, der bei jedem Protein ungefähr gleich groß ist. Demnach werden Methoden angewandt, die für Molekülgröße, Dichte oder Wasserwiderstand sensibel sind, wie die Gel-Permeations-Chromatographie, analytische Ultrazentrifuge, Kleinwinkel-Röntgenstreuung (SAXS) und Messungen des Diffusionskoeffizienten. Ungefaltete Proteine zeichnen sich durch ihren Mangel an sekundären Strukturelementen aus, die man mit fernem UV-Licht (Absorptionsmaximum bei 170–250 nm), Circulardichroismus (mit einem ausgeprägten Minimum bei ~200 nm) oder IR-Spektroskopie nachweisen könnte. Da die Peptidbindungen in der Hauptkette von ungefalteten Proteinen dem Lösungsmittel ausgesetzt sind, können sie leicht durch Proteasen gespalten werden und unterziehen sich anschließend einem Wasserstoff-Deuterium-Austausch. Dabei weisen sie eine geringe Verteilung (< 1 ppm) der 1H-NMR-Verschiebungen der Amid-Protonen auf (gefaltete Proteine zeigen typischerweise eine Verteilung von 5 ppm für ihre Amid-Protonen auf).
Neulich wurden Methoden wie die Fast parallel proteolysis (FASTpp) eingeführt, welche eine Bestimmung von gefalteten bzw. ungeordneten Anteilen des Proteins ohne vorhergehende Reinigung ermöglichen.[25][26] Selbst geringe Abweichungen in der Stabilität von Missense-Mutationen, Protein-Protein-Interaktionen und der Proteinfaltung durch (Selbst)-Polymerisation von bspw. Coiled-Coils können durch FASTpp erkannt werden, wie es kürzlich bei der Tropomyosin-Troponin-Proteininteraktion demonstriert wurde.[27] Vollständig ungeordnete Proteinregionen können aufgrund ihrer proteolytischen Anfälligkeit mit niedrigen Protease-Konzentrationen mit kleiner Expositionszeit nachgewiesen werden.[28]
Aufwendige Methoden zur Untersuchung der IDP-Struktur und -Dynamik sind zum Beispiel SAXS (Informationen über die Struktur von conformational ensembles), NMR (Feinheiten auf atomarer Ebene), Fluoreszenz (Visualisierung von Molekülinteraktionen und Konformationsänderungen), Kristallstrukturanalyse (Hervorhebung flexibler Regionen in starren Proteinkristallen), Kryoelektronenmikroskopie (Bestimmung von festen Strukturen des Proteins), dynamische Lichtstreuung (zur Darstellung der Größenverteilung von IDPs oder der Aggregationskinetik) und Circulardichroismus (zur Bestimmung von Sekundärstrukturen).
Methoden zur Untersuchung von einzelnen Molekülen von IDPs sind beispielsweise spFRET (zur Bestimmung der Konformationsflexibilität und der Kinetik von Konformationsänderungen),[29] optische Pinzetten (für hochauflösende Strukturen der conformational ensembles, Oligomere oder Aggregate von IDPs), Nanoporen (zur Bestimmung der Verteilung von Kugelformen in IDPs),[30] magnetische Pinzetten (zur Untersuchung von längerfristigen Konformationsänderungen bei geringen Krafteinwirkungen),[31] und das Rasterkraftmikroskop mit hohen Messgeschwindigkeiten (um die zeitlich-räumliche Flexibilität von IDPs zu visualisieren).[32]
Intrinsische Unordnung

Informationen zur intrinsischen Unordnung kann man aus experimentellen Daten oder mithilfe spezialisierter Software vorhersagen lassen. Algorithmen zur Vorhersage von intrinsischer Unordnung (ID) bzw. Tendenzen zur intrinsischen Unordnung können mit hoher Genauigkeit (ca. 80 %) vorausgesagt werden. Die Vorhersage basiert auf bestimmten Zusammensetzungen der Primärstruktur, Ähnlichkeiten unbestimmter Abschnitte in Datensätzen von Kristallstrukturanalysen, flexiblen Regionen bei NMR-Untersuchungen und auf physikochemische Eigenschaften der Aminosäuren.
Datenbanken
Datenbanken wurden hierfür errichtet, um zu bestimmten Proteinsequenzen Informationen zur intrinsischen Unordnung zuordnen zu können. Die Datenbank DisProt enthält eine Sammlung von Proteinsequenzen, bei denen eine intrinsische Unordnung experimentell bestimmt wurde. MobiDB ist eine Datenbank, die Informationen über die experimentell bestimmte, intrinsische Unordnung (zum Beispiel von DisProt) mit Daten aus der Kristallstrukturanalyse (über fehlende Aminosäurereste) und aus NMR-Strukturen (von flexiblen Regionen) miteinander kombiniert.
Unterscheidung von IDPs und strukturierten Proteinen
Die Trennung von geordneten und ungeordneten Proteinen ist notwendig für die Vorhersage von intrinsischer Unordnung. Dafür hat man herausgefunden, dass bestimmte Zusammensetzungen aus Aminosäuren dazu führen, dass sich eine intrinsische Unordnung bildet und bei anderen eine feste Struktur entsteht. Die hydrophilen, elektrisch geladenen Aminosäuren Alanin, Arginin, Glycin, Glutamin, Serin, Prolin, Glutaminsäure und Lysin sind Aminosäuren, welche die intrinsische Unordnung in Proteinen befördern. Dahingegen befördern die hydrophoben, ungeladenen Aminosäuren Tryptophan, Cystein, Phenylalanin, Tyrosin, Isoleucin, Valin, Leucin und Asparagin feste Strukturen (Ordnung). Die verbleibenden Aminosäuren Histidin, Methionin, Threonin und Asparaginsäure sind jeweils in geordneten und ungeordneten Regionen auffindbar.[33] Diese Information bildet die Grundlage für die meisten sequenzbasierten Vorhersagen. Regionen mit wenig oder keinen Sekundärstrukturelementen (auch bekannt als NORS, NO Regular Secondary structure)[34] sowie low-complexity regions können leicht entdeckt werden.
Vorhersagemethoden
Die Bestimmung von ungeordneten Regionen mithilfe biochemischer Methoden ist sehr kostspielig und zeitaufwendig. Da IDPs in ihrer Struktur flexibel sind, können nur bestimmte Aspekte ihrer Struktur charakterisiert werden. Somit wäre für eine vollständige Beschreibung der Struktur eine Vielzahl an verschiedenen Methoden und Experimenten nötig, sodass die Kosten zur IDP-Bestimmung astronomische Höhen erreichen. Um dieses Problem zu umgehen, entwickelte man computerbasierte Methoden, die Proteinstrukturen und -funktionen voraussagen können. Programme zur IDP-Vorhersage nutzen häufig strukturelle Informationen von Protein-Interaktionsstellen (kurze lineare Motive).[3][35] Andere Methoden zur IDP-Vorhersage beinhalten neuronale Netzwerke oder Matrizenrechnung, die auf verschiedene strukturelle und/oder biophysikalische Eigenschaften basieren.
Einige Beispiele von Softwares zur Vorhersage von IDPs sind IUPRED und Disopred. Es sollte berücksichtigt werden, dass jede Methode den Begriff „Unordnung“ anders definiert. Meta-Softwares zur Vorhersage von IDPs kombinieren verschiedene Primärsoftwares, welches die Vorhersage exakter machen könnte.[36]
Aufgrund verschiedener Methoden zur IDP-Vorhersage gestaltet sich die Messung der relativen Genauigkeit der Vorhersage als schwierig. Zum Beispiel verwenden neuronale Netzwerke eine Kombination aus verschiedenen Datensätzen. Die IDP-Vorhersage ist eine Kategorie des Gemeinschaftsexperiments CASP, das verschiedene Methoden nach ihrer Genauigkeit testet und wie genau sie Regionen in Proteinen mit fehlender 3D-Struktur auffinden können. Fehlende 3D-Strukturen werden bei PDB-Dateien als REMARK465 markiert; bei Kristallstrukturanalysen wird dies durch die fehlende Elektronendichte kenntlich gemacht.
Klinische Bedeutung
Intrinsisch ungeordnete Proteine spielen bei einigen Krankheiten eine wichtige Rolle.[37] Eine Aggregation fehlgefaltener Proteine kann zu Synucleinopathien führen. Ein Beispiel für ein intrinsisch ungeordnetes Protein ist das α-Synuclein. Aufgrund seiner strukturellen Flexibilität und seiner Anfälligkeit zur intrazellulären Modifikation kann es zu einer Aggregation und Fehlfaltung führen. Genetische Dispositionen, oxidativer und nitrosativer Stress sowie mitochondriale Beeinträchtigungen können die strukturelle Flexibilität des ungefalteten α-Synucleins beeinträchtigen und ebenfalls zu Synucleinopathien führen.[38] Viele Tumorsuppressoren besitzen eine Vielzahl an intrinsisch ungeordneten Regionen, wie p53 oder BRCA1. Diese Regionen ermöglichen erst die Interaktion mit anderen Proteinen.[39]
Computersimulationen
Aufgrund der hohen strukturellen Vielfalt von IDPs können mithilfe von NMR/SAXS-Experimenten Parameter erhalten werden, welche die Durchschnittswerte für eine Vielzahl an hochdiversen und ungeordneten Zuständen (ein conformational ensemble für ungeordnete Zustände) darstellen. Um diese Parameter, welche Informationen über die Struktur des Proteins geben, verstehen zu können, müssen genaue Darstellungen des Proteins mithilfe von Computersimulationen erstellt werden. Sogenannte all-atom molecular dynamic simulations können dafür genutzt werden, haben aber den Nachteil, dass die Genauigkeit durch Kraftfeldeinwirkungen beeinträchtigt werden. Trotzdem konnte man Kraftfelder entwickeln, die für die Untersuchung von IDPs genutzt werden können, indem die Kraftfeld-Parameter mithilfe von NMR-Daten von IDPs optimiert wurden (zum Beispiel CHARMM 22*, CHARMM 32,[40] Amberff03* und weitere).
Molekulardynamik-Simulationen, die durch experimentelle Parameter eingeschränkt werden (auch restrained-MD genannt), können auch zur Charakterisierung von IDPs verwendet werden.[41][42][43] Im Prinzip können mit MD-Simulationen (mit genauem Kraftfeld) gesamte conformational ensembles simuliert werden, solange die Simulation lange genug ausgeführt wird. Weil IDPs oftmals eine hohe strukturelle Vielfalt aufweisen, müssen diese Simulationen für eine lange Zeit mit hoher Rechenleistung laufen. Weitere Computersimulationen, welche mit kurzer Zeit laufen, sind die accelerated-MD simulations,[44] replica exchange simulations,[45][46] metadynamics,[47][48] multicanonical MD simulations[49] oder Methoden, die vereinfachte Repräsentationen des Proteins verwenden.[50]
Weiterhin kann man verschiedene Methoden und Protokolle zur Analyse von IDPs verwenden, die auf Studien zur quantitativen Untersuchung vom GC-Gehalt in Genen und deren dazugehörigen Chromosomenbanden beruhen. Mit diesen Informationen kann man Aussagen über die Funktionen von bestimmten IDP-Abschnitten treffen.[51][52]
Einzelnachweise
- Karolina Majorek, Łukasz Kozłowski, Marcin Jąkalski, Janusz M. Bujnicki: First Steps of Protein Structure Prediction. (PDF) In: John Wiley & Sons. 2008, S. 39–62. doi:10.1002/9780470741894.ch2.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner, Z. Obradovic: Intrinsically disordered protein. In: Journal of molecular graphics & modelling. Band 19, Nummer 1, 2001, S. 26–59, PMID 11381529 (Review).
- H. J. Dyson, P. E. Wright: Intrinsically unstructured proteins and their functions. In: Nature reviews. Molecular cell biology. Band 6, Nummer 3, März 2005, S. 197–208, doi:10.1038/nrm1589, PMID 15738986 (Review).
- A. K. Dunker, I. Silman, V. N. Uversky, J. L. Sussman: Function and structure of inherently disordered proteins. In: Current opinion in structural biology. Band 18, Nummer 6, Dezember 2008, S. 756–764, doi:10.1016/j.sbi.2008.10.002, PMID 18952168 (Review).
- A. Andreeva, D. Howorth, C. Chothia, E. Kulesha, A. G. Murzin: SCOP2 prototype: a new approach to protein structure mining. In: Nucleic acids research. Band 42, Januar 2014, S. D310–D314, doi:10.1093/nar/gkt1242, PMID 24293656, PMC 3964979 (freier Volltext).
- D. S. Eisenberg: How Hard It Is Seeing What Is in Front of Your Eyes. In: Cell. Band 174, Nummer 1, Juni 2018, S. 8–11, doi:10.1016/j.cell.2018.06.027, PMID 29958112.
- P. E. Wright, H. J. Dyson: Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. In: Journal of molecular biology. Band 293, Nummer 2, Oktober 1999, S. 321–331, doi:10.1006/jmbi.1999.3110, PMID 10550212 (Review).
- K. Gunasekaran, C. J. Tsai, S. Kumar, D. Zanuy, R. Nussinov: Extended disordered proteins: targeting function with less scaffold. In: Trends in biochemical sciences. Band 28, Nummer 2, Februar 2003, S. 81–85, doi:10.1016/S0968-0004(03)00003-3, PMID 12575995.
- P. Tompa: Intrinsically unstructured proteins. In: Trends in biochemical sciences. Band 27, Nummer 10, Oktober 2002, S. 527–533, PMID 12368089 (Review).
- C. J. Oldfield, B. Xue, Y. Y. Van, E. L. Ulrich, J. L. Markley, A. K. Dunker, V. N. Uversky: Utilization of protein intrinsic disorder knowledge in structural proteomics. In: Biochimica et biophysica acta. Band 1834, Nummer 2, Februar 2013, S. 487–498, doi:10.1016/j.bbapap.2012.12.003, PMID 23232152, PMC 3951342 (freier Volltext).
- T. Chouard: Structural biology: Breaking the protein rules. In: Nature. Band 471, Nummer 7337, März 2011, S. 151–153, doi:10.1038/471151a, PMID 21390105.
- M. O. Collins, L. Yu, I. Campuzano, S. G. Grant, J. S. Choudhary: Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. In: Molecular & cellular proteomics : MCP. Band 7, Nummer 7, Juli 2008, S. 1331–1348, doi:10.1074/mcp.M700564-MCP200, PMID 18388127.
- L. M. Iakoucheva, C. J. Brown, J. D. Lawson, Z. Obradović, A. K. Dunker: Intrinsic disorder in cell-signaling and cancer-associated proteins. In: Journal of molecular biology. Band 323, Nummer 3, Oktober 2002, S. 573–584, PMID 12381310.
- K. S. Sandhu: Intrinsic disorder explains diverse nuclear roles of chromatin remodeling proteins. In: Journal of molecular recognition : JMR. Band 22, Nummer 1, 2009 Jan-Feb, S. 1–8, doi:10.1002/jmr.915, PMID 18802931.
- Z. Bu, D. J. Callaway: Proteins move! Protein dynamics and long-range allostery in cell signaling. In: Advances in protein chemistry and structural biology. Band 83, 2011, S. 163–221, doi:10.1016/B978-0-12-381262-9.00005-7, PMID 21570668 (Review).
- S. H. Lee, D. H. Kim, J. J. Han, E. J. Cha, J. E. Lim, Y. J. Cho, C. Lee, K. H. Han: Understanding pre-structured motifs (PreSMos) in intrinsically unfolded proteins. In: Current protein & peptide science. Band 13, Nummer 1, Februar 2012, S. 34–54, PMID 22044148 (Review).
- A. Mohan, C. J. Oldfield u. a.: Analysis of molecular recognition features (MoRFs). In: Journal of molecular biology. Band 362, Nummer 5, Oktober 2006, S. 1043–1059, doi:10.1016/j.jmb.2006.07.087, PMID 16935303.
- K. S. Sandhu, D. Dash: Dynamic alpha-helices: conformations that do not conform. In: Proteins. Band 68, Nummer 1, Juli 2007, S. 109–122, doi:10.1002/prot.21328, PMID 17407165.
- Sarah C. Atkinson, Michelle D. Audsley, Kim G. Lieu, Glenn A. Marsh, David R. Thomas, Steven M. Heaton, Jason J. Paxman, Kylie M. Wagstaff, Ashley M. Buckle, Gregory W. Moseley, David A. Jans, Natalie A. Borg: Recognition by host nuclear transport proteins drives disorder-to-order transition in Hendra virus V. In: Scientific Reports. 8, 2018, doi:10.1038/s41598-017-18742-8.
- M. Fuxreiter: Fuzziness: linking regulation to protein dynamics. In: Molecular bioSystems. Band 8, Nummer 1, Januar 2012, S. 168–177, doi:10.1039/c1mb05234a, PMID 21927770 (Review).
- M. Fuxreiter, I. Simon, S. Bondos: Dynamic protein-DNA recognition: beyond what can be seen. In: Trends in biochemical sciences. Band 36, Nummer 8, August 2011, S. 415–423, doi:10.1016/j.tibs.2011.04.006, PMID 21620710 (Review).
- V. N. Uversky: Intrinsically disordered proteins from A to Z. In: The international journal of biochemistry & cell biology. Band 43, Nummer 8, August 2011, S. 1090–1103, doi:10.1016/j.biocel.2011.04.001, PMID 21501695 (Review).
- Kenji Sugase, H. Jane Dyson, Peter E. Wright: Mechanism of coupled folding and binding of an intrinsically disordered protein. In: Nature. 447, 2007, S. 1021, doi:10.1038/nature05858.
- Christopher J. Oldfield, A. Keith Dunker: Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. In: Annual Review of Biochemistry. 83, 2014, S. 553, doi:10.1146/annurev-biochem-072711-164947.
- D. P. Minde, M. M. Maurice, S. G. Rüdiger: Determining biophysical protein stability in lysates by a fast proteolysis assay, FASTpp. In: PloS one. Band 7, Nummer 10, 2012, S. e46147, doi:10.1371/journal.pone.0046147, PMID 23056252, PMC 3463568 (freier Volltext).
- C. Park, S. Marqusee: Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. In: Nature methods. Band 2, Nummer 3, März 2005, S. 207–212, doi:10.1038/nmeth740, PMID 15782190.
- Katarzyna Robaszkiewicz, Zofia Ostrowska, Anna Cyranka-Czaja, Joanna Moraczewska: Impaired tropomyosin–troponin interactions reduce activation of the actin thin filament. In: Biochimica et Biophysica Acta - Proteins and Proteomics. 1854, 2015, S. 381, doi:10.1016/j.bbapap.2015.01.004.
- David P. Minde, Martina Radli, Federico Forneris, Madelon M. Maurice, Stefan G. D. Rüdiger, Ashley M. Buckle: Large Extent of Disorder in Adenomatous Polyposis Coli Offers a Strategy to Guard Wnt Signalling against Point Mutations. In: PLoS ONE. 8, 2013, S. e77257, doi:10.1371/journal.pone.0077257.
- Marco Brucale, Benjamin Schuler, Bruno Samorì: Single-Molecule Studies of Intrinsically Disordered Proteins. In: Chemical Reviews. 114, 2014, S. 3281, doi:10.1021/cr400297g.
- Deanpen Japrung, Jakob Dogan, Kevin J. Freedman, Achim Nadzeyka, Sven Bauerdick, Tim Albrecht, Min Jun Kim, Per Jemth, Joshua B. Edel: Single-Molecule Studies of Intrinsically Disordered Proteins Using Solid-State Nanopores. In: Analytical Chemistry. 85, 2013, S. 2449, doi:10.1021/ac3035025.
- Duyoung Min, Kipom Kim, Changbong Hyeon, Yong Hoon Cho, Yeon-Kyun Shin, Tae-Young Yoon: Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force-generating mechanism. In: Nature Communications. 4, 2013, doi:10.1038/ncomms2692.
- Atsushi Miyagi, Yasuo Tsunaka , Takayuki Uchihashi , Kouta Mayanagi, Susumu Hirose, Kosuke Morikawa , Toshio Ando : Visualization of Intrinsically Disordered Regions of Proteins by High-Speed Atomic Force Microscopy. In: ChemPhysChem. 9, 2008, S. 1859, doi:10.1002/cphc.200800210.
- A.Keith Dunker, J.David Lawson, Celeste J Brown, Ryan M Williams, Pedro Romero, Jeong S Oh, Christopher J Oldfield, Andrew M Campen, Catherine M Ratliff, Kerry W Hipps, Juan Ausio, Mark S Nissen, Raymond Reeves, ChulHee Kang, Charles R Kissinger, Robert W Bailey, Michael D Griswold, Wah Chiu, Ethan C Garner, Zoran Obradovic: Intrinsically disordered protein. In: Journal of Molecular Graphics and Modelling. 19, 2001, S. 26, doi:10.1016/S1093-3263(00)00138-8.
- Avner Schlessinger, Christian Schaefer, Esmeralda Vicedo, Markus Schmidberger, Marco Punta, Burkhard Rost: Protein disorder—a breakthrough invention of evolution?. In: Current Opinion in Structural Biology. 21, 2011, S. 412, doi:10.1016/j.sbi.2011.03.014.
- Peter Tompa: Unstructural biology coming of age. In: Current Opinion in Structural Biology. 21, 2011, S. 419, doi:10.1016/j.sbi.2011.03.012.
- François Ferron, Sonia Longhi, Bruno Canard, David Karlin: A practical overview of protein disorder prediction methods. In: Proteins: Structure, Function, and Bioinformatics. 65, 2006, S. 1, doi:10.1002/prot.21075.
- Vladimir N. Uversky, Christopher J. Oldfield, A. Keith Dunker: Intrinsically Disordered Proteins in Human Diseases: Introducing the D Concept . In: Annual Review of Biophysics. 37, 2008, S. 215, doi:10.1146/annurev.biophys.37.032807.125924.
- Olivia Wise-Scira, Aquila Dunn, Ahmet K. Aloglu, Isin T. Sakallioglu, Orkid Coskuner: Structures of the E46K Mutant-Type α-Synuclein Protein and Impact of E46K Mutation on the Structures of the Wild-Type α-Synuclein Protein. In: ACS Chemical Neuroscience. 4, Nr. 3, 2013, ISSN 1948-7193, S. 498–508. doi:10.1021/cn3002027.
- Christopher M. Dobson: Protein folding and misfolding. In: Nature. 426, 2003, S. 884, doi:10.1038/nature02261.
- Robert B. Best, Xiao Zhu, Jihyun Shim, Pedro E. M. Lopes, Jeetain Mittal, Michael Feig, Alexander D. MacKerell: Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ and χ Dihedral Angles . In: Journal of Chemical Theory and Computation. 8, 2012, S. 3257, doi:10.1021/ct300400x.
- Robert B Best: Computational and theoretical advances in studies of intrinsically disordered proteins. In: Current Opinion in Structural Biology. 42, 2017, S. 147, doi:10.1016/j.sbi.2017.01.006.
- Song-Ho Chong, Prathit Chatterjee, Sihyun Ham: Computer Simulations of Intrinsically Disordered Proteins. In: Annual Review of Physical Chemistry. 68, 2017, S. 117, doi:10.1146/annurev-physchem-052516-050843.
- Stephen John Fox, Srinivasaraghavan Kannan: Probing the dynamics of disorder. In: Progress in Biophysics and Molecular Biology. 128, 2017, S. 57, doi:10.1016/j.pbiomolbio.2017.05.008.
- Tsuyoshi Terakawa, Shoji Takada: Multiscale Ensemble Modeling of Intrinsically Disordered Proteins: p53 N-Terminal Domain. In: Biophysical Journal. 101, 2011, S. 1450, doi:10.1016/j.bpj.2011.08.003.
- Charles K Fisher, Collin M Stultz: Constructing ensembles for intrinsically disordered proteins. In: Current Opinion in Structural Biology. 21, 2011, S. 426, doi:10.1016/j.sbi.2011.04.001.
- Alessandra Apicella, Matteo Marascio, Vincenzo Colangelo, Monica Soncini, Alfonso Gautieri, Christopher J.G. Plummer: Molecular dynamics simulations of the intrinsically disordered protein amelogenin. In: Journal of Biomolecular Structure and Dynamics. 35, 2016, S. 1813, doi:10.1080/07391102.2016.1196151.
- Gül H. Zerze, Cayla M. Miller, Daniele Granata, Jeetain Mittal: Free Energy Surface of an Intrinsically Disordered Protein: Comparison between Temperature Replica Exchange Molecular Dynamics and Bias-Exchange Metadynamics. In: Journal of Chemical Theory and Computation. 11, 2015, S. 2776, doi:10.1021/acs.jctc.5b00047.
- Daniele Granata, Fahimeh Baftizadeh, Johnny Habchi, Celine Galvagnion, Alfonso De Simone, Carlo Camilloni, Alessandro Laio, Michele Vendruscolo: The inverted free energy landscape of an intrinsically disordered peptide by simulations and experiments. In: Scientific Reports. 5, 2015, doi:10.1038/srep15449.
- Shinji Iida, Takeshi Kawabata, Kota Kasahara, Haruki Nakamura, Junichi Higo: Multimodal Structural Distribution of the p53 C-Terminal Domain upon Binding to S100B via a Generalized Ensemble Method: From Disorder to Extradisorder. In: Journal of Chemical Theory and Computation. 15, 2019, S. 2597, doi:10.1021/acs.jctc.8b01042.
- Mateusz Kurcinski, Andrzej Kolinski, Sebastian Kmiecik: Mechanism of Folding and Binding of an Intrinsically Disordered Protein As Revealed by ab Initio Simulations. In: Journal of Chemical Theory and Computation. 10, 2014, S. 2224, doi:10.1021/ct500287c.
- Vladimir N Uversky: Digested disorder. In: Intrinsically Disordered Proteins. 1, 2014, S. e25496, doi:10.4161/idp.25496.
- Susan Costantini, Ankush Sharma, Raffaele Raucci, Maria Costantini, Ida Autiero, Giovanni Colonna: Genealogy of an ancient protein family: the Sirtuins, a family of disordered members. In: BMC Evolutionary Biology. 13, 2013, S. 60, doi:10.1186/1471-2148-13-60.