Helmchen-Synthese
Die Helmchen-Synthese ist eine Namensreaktion aus dem Bereich der Organischen Chemie. Sie wird dazu benutzt α-chirale Carbonsäuren beziehungsweise β-chirale Alkohole zu synthetisieren. Die Reaktion ist nach ihrem Entwickler, dem deutschen Chemiker Günter Helmchen benannt.[1][2]
Helmchen-Auxiliar
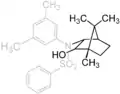
Die Helmchen-Synthese bedient sich eines chiralen Auxiliars, das auch als Helmchen-Auxiliar bezeichnet wird. Dieses ist in mehreren Reaktionsstufen aus kommerziell erhältlichem Campher zugänglich. Da beide Enantiomere des Camphers erhältlich sind können auf diesem Wege auch beide Enantiomere des Auxiliars erhalten werden. Das Auxiliar besitzt sterisch anspruchsvolle Phenyl- und Dimethylphenylreste. Diese bewirken, dass bei einer Reaktion eine Seite des Moleküls effektiv abgeschirmt ist und eine Reaktion nur im zweiten Halbraum stattfinden kann. Dies bildet die Basis der Enantioselektivität, die dieses Auxiliar induziert. Durch Veresterung kann das Auxiliar an die Carbonsäure, an der die Reaktion stattfinden soll, gekuppelt werden.[1][2]
Reaktionsmechanismus
Der vorliegende Ester ist eine CH-acide Verbindung, die im ersten Schritt mit einer Base deprotoniert wird, wobei ein Enolat gebildet wird. Als Basen eignen sich Lithiumamide wie Lithiumdiisopropylamid (LDA). Bessere Ergebnisse werden jedoch mit Amidbasen erhalten, die einen sterisch anspruchsvolleren Rest als die Isopropylgruppe tragen, beispielsweise mit Lithiumcyclohexylisopropylamid. Wird lediglich die Base zur Enolatbildung eingesetzt, wird gemäß dem Ireland-Modell das trans-Enolat erhalten, bei Zugabe von Hexamethylphosphorsäuretriamid (HMPT) wird das cis-Enolat erzeugt, da das HMPT eine Komplexierung der lithiumhaltigen Base mit dem Enolat während des Übergangszustandes verhindert. Dieses wird nun mit der zu kuppelnden Verbindung zur Reaktion gebracht. Hierzu muss diese Verbindung eine Abgangsgruppe tragen, welche die Verbindung beim nukleophilen Angriff des Enolats im Sinne einer Substitutionsreaktion verlassen kann. Hierzu eignen sich beispielsweise die meist gut zugänglichen Bromide. In diesem Schritt wird ein in α-Position substituierter Ester mit hoher Diastereoselektivität erhalten. Das reine Diastereomer kann durch anschließende chromatographische Aufarbeitung erhalten werden. Eine darauf folgende Reduktion mit Lithiumaluminiumhydrid (LAH) führt zur Bildung des gewünschten chiralen Alkohols unter Abspaltung des Auxiliars.[1]
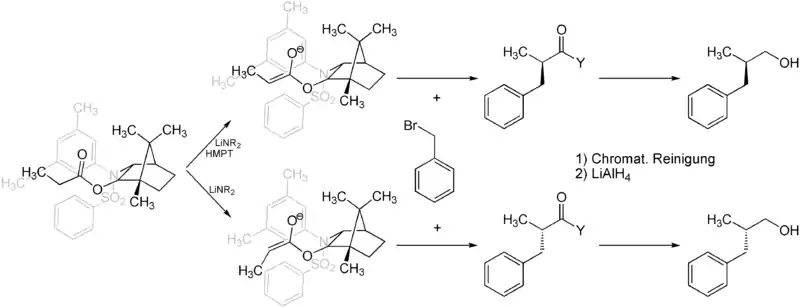
Mit Hilfe des Helmchen-Auxiliars gelang beispielsweise die enantioselektive Synthese der α-Tocopherol-Seitenkette.[3]
Quellen
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 549–552.
- M. Christmann, S. Bräse, D. Seebach: Asymmetric Synthesis. 2. Auflage, Wiley-VCH, Weinheim, 2007, ISBN 978-3-527-32093-6, S. 3–9.
- Günter Helmchen, Roland Schmierer: A Total Synthesis of Enantiomerically Pure Vitamin E Side Chain Using a Chiral Propionate Synthon. In: Tetrahedron Letters. Band 24, Nr. 12, 1983, S. 1235–1238, doi:10.1016/S0040-4039(00)81623-8.